
Abstract
Colon adenocarcinoma (COAD) is a leading cause of cancer-related mortality worldwide, with immune escape being a significant factor in the failure of immunotherapy. This study investigates the correlation between Immunosurveillance-related genes and the prognosis of COAD patients, utilizing data from 1140 patients across four public databases: The Cancer Genome Atlas (TCGA), International Cancer Genome Consortium (ICGC), Array-express, and Gene Expression Omnibus (GEO). Employing Cox regression analysis, we identified 182 immune genes significantly linked to overall survival (OS) and established an Immunosurveillance score (ISs) based on 16 of these genes. The ISs score was validated using independent datasets, revealing that patients in the high-ISs group exhibited significantly poorer OS compared to those in the low-ISs group, as demonstrated by Kaplan–Meier curves and Cox regression analyses. Moreover, the ISs score showed a negative correlation with immune scores across multiple datasets. Notably, a higher ISs score was associated with improved recurrence-free survival (RFS) and OS in patients treated with PD-1 and CTLA4 inhibitors. Our findings suggest that the ISs score, derived from Immunosurveillance-related genes, has the potential to serve as a valuable prognostic marker and a tool for identifying COAD patients who may benefit from immunotherapy in clinical settings.
Similar content being viewed by others

Introduction
COAD is one of the most frequent malignant lesions of the digestive system in humans, with an insidious onset. Although COAD has a relatively low incidence worldwide, it ranks the fourth leading cause of cancer-related death in the world1,2,3. Radical surgery is the only effective treatment for the cure of COAD. Unfortunately, most COAD patients are present in advanced stages when they are diagnosed and not amenable for surgery4,5,6. Even when COADs undergo surgery, the 5-year postoperative survival rate is still not ideal. Despite great advances in chemotherapy for colon cancer, chemo-resistance and toxic side effects also inhibited the improvement of COAD patients’ prognosis after treatment7,8. Previous studies have demonstrated the 5-year survival rate of COAD is less than 9%, which is far lower than that of other malignancies2,9.
Recent years, besides chemotherapy, immunotherapy has become a research hotspot in the treatment of malignances, included colon cancer. However, it has a poor effect on pancreatic cancer with PD-1/PD-L1 blockade monotherapy3,7. Immune evasion might be the main cause of immunotherapy failure, which reflected in the resistance to immune checkpoint blockade (ICB) therapy10,11. More and more studies have disclosed the molecular mechanisms of immune evasion in colon cancer, which increases the difficulty in the treatment of COAD. Considering the lethal nature and limited efficacy of immunotherapy, the prognosis of COAD patients should be assessed holistically. Surgeons should evaluate treatment benefits using immune-related gene markers alongside other factors, including clinicopathological features (TNM staging, differentiation, invasion depth), molecular characteristics (MSI status, RAS/BRAF mutations), surgical factors (margin status, resection extent), and patient factors (age, comorbidities, nutritional status)12.
In this study, we developed and validated an Immunosurveillance score (ISs) with the whole genome expression data from several datasets for COAD. More importantly, the IEGPI score could identify COAD patients with an unfavorable prognostic outcome after surgery, and COAD patients with a high sensitivity to immunotherapy after received with both PD-1 and CTLA413,14.
Results
Immune surveillance-related genes linked to prognosis in COAD
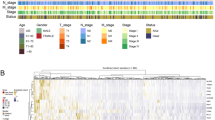
In normal mammalian systems, immune cells constantly monitor the physiological state of their microenvironment, with CTLs identifying and eliminating malignant cells. According to existing literature, 182 genes have been identified that are associated with the escape of cancer cells from T-cell surveillance and eradication (Supplementary Table S1). To investigate whether these genes affect the OS of colorectal cancer patients, we performed a COX regression analysis on the transcriptome levels of these 182 genes, comparing them with the survival times of the corresponding samples. This analysis resulted in 25 genes whose mRNA expression levels significantly impacted OS (Supplementary Table S2). Through the integration of five gene sets, we eventually identified 16 critical genes (Supplementary Table S3 and Fig. 1A). Among these, high expression of six genes was associated with favorable prognosis, while high expression of the remaining ten was linked to poor patient survival (Fig. 1B).

Illustrates the construction of the IS scoring model. (A) Expression levels of risk genes across five independent datasets. (B) The impact of risk genes on patient prognosis. Orange represents risk genes, where high expression correlates with shorter survival time. Blue represents protective genes, where low expression correlates with shorter survival time. (C,D) Functional annotation of genes associated with colon cancer survival. (E) Heatmap of the distribution of clinical features for each sample. (F) Single-cell type annotation in colon cancer. (G) IS groupings for each cell type. (H) IS scores for various cell types.
KEGG annotation analysis indicated that the risk-associated genes were involved in pathways related to autophagy, the TNF signaling pathway, and GPI anchor biosynthesis (Fig. 1C). GO annotation analysis revealed that these risk genes primarily participated in biological processes related to post-transcriptional silencing and the inhibition of protein translation (Fig. 1D). To assess the representativeness of the data and potential biases, we further visualized the differences in patient characteristics across the four public databases. The heatmap is divided into four sections: Immune Surveillance Score, Overall Event, Gender, and Age (Fig. 1E). Further investigation into single-cell transcriptome data allowed us to identify cell populations such as B cells, epithelial cells, mast cells, myeloid cells, stromal cells, and T cells (Fig. 1F). Notably, epithelial cells had the lowest ISs, while T cells displayed the highest ISs (Fig. 1G,H). Thus, these findings suggest that certain immune surveillance-related genes have a significant impact on the prognosis of colorectal cancer patients, with implications for potential therapeutic targets and prognostic indicators.
Patients with high IS tend to have poorer outcomes
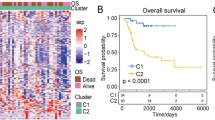
In colorectal cancer patients, the transcriptional expression of each risk gene is strongly correlated with ISs, and this pattern holds consistently across all datasets (Fig. 2A). Microarray sequencing data were used as the training set for model development, and we found that patients with high IS scores had shorter overall survival compared to those with low IS scores (Fig. 2B). This trend was also observed in the TCGA-COAD dataset, with similar results (Fig. 2C). To explore whether patients’ IS scores could indicate the activation or suppression of tumor-related pathways, we calculated the correlation between the average scores for Hallmark pathways and IS scores in each dataset. The results showed significant variability in the correlation between pathways and IS scores across different datasets (Fig. 2D). In addition, we further explored the interactions between different subsets and constructed a communication network diagram among the various cell subsets, including the Number of interactions and Interaction weights/strength (Fig. 2E).

Evaluation of Patient Survival Prognosis Based on IS Score. (A) The correlation of each gene in the computation of the IS score. (B,C) Kaplan–Meier analysis of the overall survival of patients stratified by the median IS score to assess the relationship between overall survival and IS score. (D) The relationship between IS score and the activation status of transcriptional pathways in the samples. (E) Communication network diagram of the different cell subsets.
ISs are an independent prognostic factor in colon cancer patients
Through multivariate Cox regression analysis, we found that ISs could serve as an independent predictor for overall prognosis in patients with colon cancer (Fig. 3A, P = 0.0171). Additionally, Kaplan–Meier (K-M) analyses of three independent datasets revealed that patients with high immune scores (HIS) had significantly shorter progression-free intervals compared to those with low immune scores (LIS) (Fig. 3B,C). The specific datasets are GSE17538, GSE38832, and GSE39582. To assess its advantages and limitations in terms of predictive accuracy, stability, and clinical practicability, we compared the AUC (Area Under the Curve) values of our immune surveillance score (IS) model with those of other published prognostic models for colon adenocarcinoma. As shown in Fig. 3D, our model achieved an AUC of 0.82, significantly higher than the AUC values reported in previous studies. This highlights the superior predictive accuracy of the IS score model, indicating that our approach offers more reliable prognostic power in evaluating patient outcomes.

The IS score is an independent prognostic factor for patients with colon cancer. (A) COX multivariate analysis of factors influencing overall survival. (B) Kaplan–Meier analysis of disease-free survival based on patient stratification by the median IS score. (C) The differences in ISs were compared between patients with relapse and those in remission. (D) Bar chart comparing the AUC values of the IS score model with those of other research models.
Genomic landscape differences between HIS and LIS patients
The accumulation of somatic gene mutations in body cells is a crucial factor in the development of cancer. In order to explore the relationship between ISs and gene mutations, we first identified a positive correlation between ISs and TMB in the TCGA-COAD dataset (Fig. 4A). Additionally, among the six genes with the highest mutation frequencies in colon cancer patients, namely APC, TP53, TTN, KRAS, SYNE1, and PIK3CA (Fig. 4B), the expression levels of APC, KRAS, and PIK3CA were positively correlated with ISs, while TP53 transcriptional expression levels showed a negative correlation (Fig. 4C). Furthermore, we observed a negative correlation between ISs and Copy Number Variation (CNV) scores (Fig. 4D). Subsequently, we compared the genes exhibiting the greatest differences in mutations between HIS and LIS patients, which included BRAF, AHNAK2, KMT2B, FMN2, KMT2D, DST, SLIT3, KIAA1549L, CDON, and SLC4A7 (Fig. 4E). Furthermore, we compared the differences in mutation sites of the same genes between HIS and LIS patients (Fig. 4F–H, BRAF, KMT2B, and AHNAK2), revealing a significantly higher number of mutation sites in KMT2B among HIS patients, with no significant differences observed in other genes. Moreover, we conducted a comprehensive analysis of chromosomal amplifications and deletions in each patient. While both HIS and LIS patients exhibited concurrent 16p13.3 deletions, LIS patients displayed high-frequency amplifications at 8p11.23, 13q12.13, and 20q12.21, and high-frequency deletions at 4q22.1 and 20p12.1. Conversely, HIS patients showed high-frequency amplifications at 8q24.21 and 11p15.1, along with high-frequency deletions at 1p33, 4q22.1, and 6q26 (Fig. 4I,J).

Relationship between IS score and genomic events in colon cancer patients. (A) The correlation between ISs and TMB scores. (B) The figure depicts the frequency of gene mutations in TCGA-COAD. (C) The analysis presents the correlation between the expression of high mutation frequency genes and ISs. (D) The analysis demonstrates the correlation between CNV scores and ISs. (E) The disparity in gene mutation frequency between HIS and LIS patients. (F–H) The comparison focuses on the gene mutation sites between HIS and LIS patients, specifically analyzing BRAF, KMT2B, and AHNAK2. (I,J) The analysis compares chromosomal amplifications and deletions between LIS and HIS patients.
The response rates to immunotherapy are low in HIS patients
The infiltration of immune cells within tumor tissues significantly impacts both the survival and proliferation of tumor cells, concurrently serving as a pivotal determinant of the efficacy of immunotherapeutic interventions. Leveraging the ESTIMATE algorithm, we estimated the tumor purity and immune cell infiltration scores at the transcriptomic level for each sample. Strikingly, across the majority of datasets, we observed a positive correlation between ISs and tumor purity, juxtaposed with a negative correlation with immune cell infiltration levels (Fig. 5A,B). Further analysis revealed a compelling trend: higher ISs corresponded to decreased infiltration of plasmacytoid dendritic cells (pDCs) and heightened levels of neutrophils and M2 macrophages across the five datasets (Fig. 5C). Interestingly, the expression patterns of immune checkpoint-related genes exhibited considerable variability across these datasets, with ICOSLG demonstrating a positive correlation with ISs (Fig. 5D).

Relationship between IS scores and the extent of immune cell infiltration in the tumor tissues of patients. (A) Correlation between tumor purity and IS scores in five independent datasets for colorectal cancer. (B) Correlation between the immune infiltration score and the IS score for each sample. (C) Relationship between the infiltration levels of 28 types of immune cells and IS scores. (D) Relationship between the expression of immune checkpoint-related genes and IS scores.
We opted to validate whether ISs could serve as predictive tools for the efficacy of ICI therapy in cancer patients, utilizing an immunotherapy cohort collected from TIED study. In melanoma and gastric cancer patients receiving both PD1 and CTLA4 inhibitors, ISs effectively discerned treatment response states (Fig. 6A–E, AUC = 0.727, 0.766, 0.644, 0.773, 0.647). Among melanoma patients treated with PD1 inhibitors, HIS individuals exhibited shorter OS and RFS compared to LIS individuals (Fig. 6F,G, P = 0.085, P = 0.05), a similar trend observed in urothelial carcinoma patients (Fig. 6H, P = 0.014). Notably, ISs scores were significantly higher in non-responsive patients compared to responders (Fig. 6I). Furthermore, employing TIED to predict response status post-immunotherapy in colorectal cancer patients, we found that HIS patients displayed significantly elevated CAF scores, T cell exclusion scores, and TIED scores compared to LIS group, indicating diminished benefits from immunotherapy in HIS patients (Fig. 6J–L).

The predictive capacity of the IS scores in assessing the response of cancer patients to immunotherapy. (A) The study demonstrates the use of PD-1 inhibitors in melanoma patients. (B) The study depicts the combined use of PD-1 inhibitors and CTLA-4 inhibitors in melanoma patients. (C). The study displays the use of PD-1 inhibitors in gastric cancer patients. (D) The study demonstrates the use of CTLA-4 inhibitors in melanoma patients. (E) The study represents the application of ACT therapy in melanoma patients. (F–H) Segment patients into groups based on the median IS score for Kaplan–Meier analysis to compare overall survival (OS) and progression-free survival (PFS) between the groups. (F,G) Focus on the use of PD-1 inhibitors alone in the treatment of melanoma. (H) The study shows the use of PD1 inhibitors in a cohort of urothelial carcinoma. (I) Compare the IS score between responders and non-responders to immunotherapy in urothelial carcinoma (P = 0.02). (J–L) Examine the relationship between the IS score and levels of response to immunotherapy across three independent transcriptomic datasets of colon cancer.
Validation of high-risk gene expression in colon adenocarcinoma
To further validate the expression of the high-risk genes associated with colon adenocarcinoma prognosis, we performed immunohistochemistry (IHC) and Western blot (WB) experiments. First, we utilized the Human Protein Atlas (HPA) database to obtain publicly available IHC images of colon adenocarcinoma tissues. We selected four high-risk genes—MAP3K7, HEXIM1, HCFC2, and DICER1—for experimental validation. The IHC results demonstrated significantly higher protein expression levels of these genes in tumor tissues compared to normal tissues. ImageJ software was used to quantify the average optical density (AOD), confirming a significant increase in expression in tumor samples (Fig. 7A–H).

Validation of high-risk gene expression in colon adenocarcinoma. (A,C,E,G) Immunohistochemistry (IHC) images from the HPA database showing the expression of MAP3K7, HEXIM1, HCFC2, and DICER1 in normal and tumor tissues. The tumor tissues exhibit significantly higher staining intensity, indicating elevated expression of these genes. Scale bar = 100 µm. (B,D,F,H) Quantification of IHC staining using ImageJ software reveals significantly higher average optical density (AOD) of MAP3K7, HEXIM1, HCFC2, and DICER1 in tumor tissues compared to normal tissues. (I) Western blot (WB) analysis confirms the overexpression of these genes in tumor tissues compared to normal tissues, with GAPDH used as a loading control. (J) Quantification of WB results shows the relative expression levels of MAP3K7, HEXIM1, HCFC2, and DICER1 in colon adenocarcinoma, with band intensities measured using ImageJ and normalized to GAPDH. The tumor tissues exhibit significantly higher protein expression compared to normal tissues.
To further verify the differential expression of these genes at the protein level, we conducted WB analysis on paired normal and tumor tissues. The results revealed notably higher protein expression of MAP3K7, HEXIM1, HCFC2, and DICER1 in tumor tissues (Fig. 7I). Quantification of the WB results using ImageJ showed significantly higher relative expression levels of these genes in tumor tissues compared to normal tissues, with band intensities normalized to GAPDH (Fig. 7J). These findings provide experimental evidence supporting the bioinformatics analysis, confirming the elevated expression of these high-risk genes in colon adenocarcinoma. This validation further strengthens their potential role as prognostic markers and therapeutic targets in colon cancer.
Discussion
This study proposes the immune surveillance score (ISs) as an effective tool for prognostic evaluation and prediction of immunotherapy responses in colorectal cancer (COAD) patients. By analyzing transcriptomic data of 182 immune surveillance-related genes, a scoring model based on 27 genes was developed, effectively predicting patients’ overall survival (OS) and recurrence-free survival (RFS). Patients with high ISs scores typically exhibited poorer prognosis and lower levels of immune cell infiltration9,13,15. Further analysis revealed that high ISs scores were positively correlated with tumor mutation burden (TMB) and could effectively predict responses to immune checkpoint inhibitors (e.g., PD1 and CTLA4)16,17,18,19,20. This suggests that the ISs score can serve not only as a predictive biomarker for immunotherapy responses but also as a tool to assess immune evasion mechanisms, thereby guiding the development of personalized treatment plans.
The innovation of this study lies in introducing the immune surveillance score (ISs) as a novel prognostic evaluation tool and demonstrating its potential in immunotherapy. The ISs score integrates the expression levels of multiple immune surveillance-related genes, comprehensively reflecting changes in the tumor immune microenvironment, thus providing more precise information for clinical treatment21,22,23. Unlike existing single-gene biomarkers, the ISs score evaluates multiple factors, including immune cell infiltration, tumor mutation burden, and immune evasion mechanisms, offering a more comprehensive prediction of patients’ responses to immune checkpoint inhibitors9,13,15. This offers a new direction for personalized decision-making in immunotherapy, especially in colorectal cancer, where immune evasion mechanisms are prominent, highlighting the significant clinical value of the ISs score24,25,26.
The initial hypothesis of this study was that the expression of immune surveillance-related genes is closely associated with the prognosis of colorectal cancer (COAD) patients and that the ISs score can serve as an important tool for predicting immunotherapy responses27,28. Through data analysis, we found that high ISs scores were closely associated with poorer survival, lower levels of immune cell infiltration, and higher tumor mutation burden, which are linked to the presence of immune evasion mechanisms16. Particularly in immunotherapy, patients with high ISs scores generally exhibited poor responses, validating the hypothesis that ISs serves as a predictor of immunotherapy sensitivity9,13,15. Furthermore, the strong correlations between the ISs score, tumor gene mutation frequency, and the immune microenvironment further support the potential application of this model in immunotherapy6,29.
Despite providing an important theoretical basis for the application of the ISs score in colorectal cancer, this study has certain limitations. First, the research data were primarily derived from public databases, which may introduce selection bias and inconsistencies in data quality. Although we validated the effectiveness of the ISs score across multi-center datasets, data heterogeneity might affect its generalizability9,13. Second, the ISs score was constructed based on transcriptomic data, which may not fully reflect protein-level expression. Future studies should validate its accuracy and reliability using more direct experimental data6. Additionally, this study primarily focused on colorectal cancer; future research should extend to other types of cancer to evaluate the cross-cancer applicability and predictive capabilities of the ISs score in immunotherapy responses16.
To facilitate the translation and application of the findings from this study, several key measures need to be taken. First, the clinical application of the ISs score requires broader clinical validation, particularly through multi-center, large-sample studies30,31,32. These studies should incorporate techniques such as immunohistochemistry and single-cell RNA sequencing to further evaluate the ISs score’s ability to predict prognosis and immunotherapy response in various clinical contexts17,24,25. Second, the ISs score should be integrated with existing clinical diagnostic and treatment protocols, including combining it with patient clinical characteristics, tumor staging, and other biomarkers, to enhance its applicability in personalized treatment6. Additionally, the ISs score can be combined with liquid biopsy technologies to develop it into a convenient and non-invasive tool for predicting immunotherapy responses33,34. Finally, to ensure the clinical utility of the ISs score, researchers need to collaborate with pharmaceutical companies and clinicians to conduct relevant clinical trials, evaluating its practical applications in various immunotherapy regimens and further advancing the development of precision medicine16,17,18,19.
Future research directions should delve deeper into the biological mechanisms of the immune surveillance score, particularly its relationship with immune evasion and immune cell infiltration33. Through single-cell technologies and high-throughput sequencing, further exploration is needed to elucidate the complex interactions between the ISs score, immune cell subpopulations, and the tumor microenvironment9,11. Moreover, the ISs score could be extended to other cancer types, such as lung cancer, gastric cancer, and melanoma, to assess its prognostic and immunotherapy response predictive capabilities across different tumor types16. As immunotherapy targets continue to evolve, future research could also explore combining the ISs score with new immunotherapy targets to further improve treatment efficacy and patient survival24,25.
In conclusion, the immune surveillance score (ISs) has significant clinical potential as a predictive and prognostic tool for immunotherapy response and prognosis in colorectal cancer. With further clinical validation and cross-cancer studies, the ISs score is expected to become a key indicator in tumor immunotherapy, driving the development of precision medicine and personalized treatment17,18,19.
Methods
Data source
We collected six sets of RNA microarray sequencing data for colon cancer from the GEO database, including GSE39582 (n = 536), GSE17538 (n = 288), GSE71729 (n = 125), GSE57495 (n = 63), and GSE62452 (n = 66), along with the COAD data from the TCGA database (n = 446). Additionally, the data for immunotherapy cohorts were obtained from Tumor immune dysfunction and exclusion (TIDE, http://tide.dfci.harvard.edu/).
Identification of prognostic genes for survival
Cytotoxic T lymphocytes (CTL) play a crucial role in the immune system’s direct action to destroy cancer cells. According to the literature, 182 genes are involved in this process, collectively referred to as immune surveillance-related genes (Supplementary Table S1). We analyzed the expression profiles of these 182 genes in our collected dataset and stratified each sample into high- and low-expression groups. This stratification formed the basis for correlating overall survival in colorectal cancer patients, employing Cox regression analysis to identify risk genes with significant impact on survival duration. To further identify generally applicable key genes, we consolidated the results from five datasets using a fixed-effects model. This meta-analysis approach allowed us to isolate and validate genes consistently associated with survival outcomes across different datasets.
Develop an immuno-surveillance-related gene signature for prognosis
To create a prognostic gene signature associated with immune surveillance, each identified risk gene’s standard estimate (SE) and hazard ratio (HR) were used as weights reflecting its impact on patient survival. These weights were then multiplied by the gene expression levels to compute the ISs for each patient.
Assessment of tumor immune scores
The ESTIMATE algorithm is used to determine immune scores for colorectal cancer samples, providing an estimate of immune infiltration within tumor tissue. A higher estimate score indicates a greater proportion of tumor cells within the sample, suggesting lower immune infiltration.
Estimation of immune cell infiltration
Using the ESTIMATE algorithm, we quantify the infiltration of 28 types of immune cells within tumor tissues, based on the transcriptomic expression levels for each sample. A higher score indicates a greater degree of immune cell infiltration.
Tumor mutational burden (TMB) assessment
To evaluate TMB, exome sequencing MAF (Mutation Annotation Format) files for colorectal cancer were downloaded from the TCGA database. These files were imported into an R environment using the maftools package, and TMB was calculated with the tmb function. Additionally, a mutation landscape for each sample was plotted.
Gene functional enrichment analysis
For the identified risk genes, functional annotation analysis was performed using the ClusterProfiler package in conjunction with the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) databases. Furthermore, the Gene Set Enrichment Analysis (GSEA) algorithm was employed to determine the level of pathway enrichment at the transcriptomic level among patients with different immune scores.
Utilization of the human protein atlas database
The Human Protein Atlas (HPA) is an open-access database that provides comprehensive information on protein expression across human tissues and cells. We used the HPA to examine the expression of MAP3K7, HEXIM1, HCFC2, and DICER1 in colon adenocarcinoma tissues through immunohistochemistry (IHC) images.
Western blot
Proteins were separated by SDS-PAGE, loading 10–20 μg of protein per lane, and electrophoresis was performed at 80 V for 30 min, followed by 120 V until dye migration. Proteins were transferred to NC membranes using a wet transfer system (350 mA for 90 min) in Tris–glycine buffer. The membranes were blocked with 10% skim milk in TBST for 1 h, then incubated overnight at 4°C with primary antibodies against MAP3K7, HEXIM1, HCFC2, and DICER1 at dilutions of 1:1000, 1:1000, 1:1000, and 1:250, respectively. After washing with TBST, the membranes were incubated with HRP-conjugated secondary antibody (1:10,000) for 1 h at room temperature. Protein signals were detected using an ECL substrate (1:1 reagent A: B mixture) and quantified with ImageJ by normalizing the target band intensities to the loading controls.
Statistical analysis
Continuous variables were analyzed using either the t-test or the Wilcoxon rank-sum test, while categorical variables were compared using either the chi-squared test or Fisher’s exact test. In survival analysis, the log-rank test was used to compare overall survival rates between two groups. Statistical significance was defined as a P-value < 0.05.
Data availability
Publicly available datasets were analyzed in this study. The single cell RNA-seq datasets used for cell states analysis were all downloaded from GEO database (https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE39582, GSE17538, GSE71729, GSE57495 and GSE62452, and from the UCSC Xena platform (https://xenabrowser.net/) under accession TCGA Colon adenocarcinoma (COAD). Additionally, the data for immunotherapy cohorts were obtained from TIDE (http://tide.dfci.harvard.edu/).
References
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480. https://doi.org/10.1016/S0140-6736(19)32319-0 (2019).
Lee, H.-O. et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594–603. https://doi.org/10.1038/s41588-020-0636-z (2020).
Le, D. T. et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol. 38, 11–19. https://doi.org/10.1200/JCO.19.02107 (2020).
Lu, W. et al. Reprogramming immunosuppressive myeloid cells facilitates immunotherapy for colorectal cancer. EMBO Mol. Med. 13, e12798. https://doi.org/10.15252/emmm.202012798 (2021).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13, 816. https://doi.org/10.1038/s41467-022-28421-6 (2022).
Morton, D. et al. Preoperative chemotherapy for operable colon cancer: Mature results of an international randomized controlled trial. J. Clin. Oncol. 41, 1541–1552. https://doi.org/10.1200/JCO.22.00046 (2023).
Jorissen, R. N. et al. Metastasis-associated gene expression changes predict poor outcomes in patients with dukes stage B and C COLORECTAL Cancer. Clin. Cancer Res. 15, 7642–7651. https://doi.org/10.1158/1078-0432.CCR-09-1431 (2009).
Foxtrot Collaborative Group, null. Feasibility of preoperative chemotherapy for locally advanced, operable colon cancer: the pilot phase of a randomised controlled trial. Lancet Oncol. 13, 1152–1160 (2012). https://doi.org/10.1016/S1470-2045(12)70348-0.
Samstein, R. M. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206. https://doi.org/10.1038/s41588-018-0312-8 (2019).
Floudas, C. S. et al. A pilot study of the PD-1 targeting agent AMP-224 used with low-dose cyclophosphamide and stereotactic body radiation therapy in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 18, e349–e360. https://doi.org/10.1016/j.clcc.2019.06.004 (2019).
Ganesh, K. et al. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 16, 361–375. https://doi.org/10.1038/s41575-019-0126-x (2019).
Fiegle, E. et al. Dual CTLA-4 and PD-L1 blockade inhibits tumor growth and liver metastasis in a highly aggressive orthotopic mouse model of colon cancer. Neoplasia 21, 932–944. https://doi.org/10.1016/j.neo.2019.07.006 (2019).
Tintelnot, J. & Stein, A. Immunotherapy in colorectal cancer: Available clinical evidence, challenges and novel approaches. World J. Gastroenterol. 25, 3920–3928. https://doi.org/10.3748/wjg.v25.i29.3920 (2019).
Zhang, Y. et al. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett. 470, 84–94. https://doi.org/10.1016/j.canlet.2019.10.016 (2020).
Wang, Y. et al. Combination of fruquintinib and anti-PD-1 for the treatment of colorectal cancer. J. Immunol. 205, 2905–2915. https://doi.org/10.4049/jimmunol.2000463 (2020).
Shin, A. E., Giancotti, F. G. & Rustgi, A. K. Metastatic colorectal cancer: Mechanisms and emerging therapeutics. Trends Pharmacol. Sci. 44, 222–236. https://doi.org/10.1016/j.tips.2023.01.003 (2023).
Hu, G. et al. The anoikis-related gene signature predicts survival accurately in colon adenocarcinoma. Sci. Rep. 13, 13919. https://doi.org/10.1038/s41598-023-40907-x (2023).
Lin, K. X. et al. PD-1 and PD-L1 inhibitors in cold colorectal cancer: challenges and strategies. Cancer Immunol. Immunother. 72, 3875–3893. https://doi.org/10.1007/s00262-023-03520-5 (2023).
Xu, J., Yang, J., Pan, X. & Wang, J. Prognostic and immunotherapeutic significance of immunogenic cell death-related genes in colon adenocarcinoma patients. Sci. Rep. 13, 19188. https://doi.org/10.1038/s41598-023-46675-y (2023).
Kim, C. G. et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci. Immunol. 4, eaay0555. https://doi.org/10.1126/sciimmunol.aay0555 (2019).
Idos, G. E. et al. The prognostic implications of tumor infiltrating lymphocytes in colorectal cancer: A systematic review and meta-analysis. Sci. Rep. 10, 3360. https://doi.org/10.1038/s41598-020-60255-4 (2020).
Cai, X. et al. Apatinib enhanced anti-PD-1 therapy for colon cancer in mice via promoting PD-L1 expression. Int. Immunopharmacol. 88, 106858. https://doi.org/10.1016/j.intimp.2020.106858 (2020).
Monjazeb, A. M. et al. A randomized trial of combined PD-L1 and CTLA-4 inhibition with targeted low-dose or hypofractionated radiation for patients with metastatic colorectal cancer. Clin Cancer Res. 27, 2470–2480. https://doi.org/10.1158/1078-0432.CCR-20-4632 (2021).
Guil-Luna, S. et al. Association of tumor budding with immune evasion pathways in primary colorectal cancer and patient-derived xenografts. Front. Med. (Lausanne) 7, 264. https://doi.org/10.3389/fmed.2020.00264 (2020).
Luo, J. et al. Establishment of an immune-related gene pair model to predict colon adenocarcinoma prognosis. BMC Cancer 20, 1071. https://doi.org/10.1186/s12885-020-07532-7 (2020).
Miao, Y., Wang, J., Ma, X., Yang, Y. & Mi, D. Identification prognosis-associated immune genes in colon adenocarcinoma. Biosci. Rep. 40, 1734 (2020).
Liu, Z. et al. Clinical significance and inflammatory landscape of anovel recurrence-associated immune signature in stage ii/iii colorectal cancer. Front. Immunol. 12, 702594. https://doi.org/10.3389/fimmu.2021.702594 (2021).
Doleschel, D. et al. Regorafenib enhances anti-PD1 immunotherapy efficacy in murine colorectal cancers and their combination prevents tumor regrowth. J. Exp. Clin. Cancer Res. 40, 288. https://doi.org/10.1186/s13046-021-02043-0 (2021).
Luo, Q., Zhou, P., Chang, S., Huang, Z. & Zeng, X. Construction and validation of a prognostic model for colon adenocarcinoma based on bile acid metabolism-related genes. Sci. Rep. 13, 12728. https://doi.org/10.1038/s41598-023-40020-z (2023).
Stefani, C. et al. Growth factors, PI3K/AKT/mTOR and MAPK signaling pathways in colorectal cancer pathogenesis: Where are we now?. Int. J. Mol. Sci. 22, 10260. https://doi.org/10.3390/ijms221910260 (2021).
Lu, H. et al. The immune escape signature predicts the prognosis and immunotherapy sensitivity for pancreatic ductal adenocarcinoma. Front. Oncol. 12, 978921. https://doi.org/10.3389/fonc.2022.978921 (2022).
Yan, H., Talty, R. & Johnson, C. H. Targeting ferroptosis to treat colorectal cancer. Trends Cell Biol. 33, 185–188. https://doi.org/10.1016/j.tcb.2022.11.003 (2023).
Jiang, Z., Xu, J., Zhang, S., Lan, H. & Bao, Y. A pairwise immune gene model for predicting overall survival and stratifying subtypes of colon adenocarcinoma. J. Cancer Res. Clin. Oncol. 149, 10813–10829. https://doi.org/10.1007/s00432-023-04957-y (2023).
Huang, X. et al. Case report: A clinical report of photodynamic neoadjuvant combined with fluorescent laparoscopic localization robotic surgery for the treatment of patients with advanced colorectal cancer combined with obstruction. Front. Immunol. 15, 1403613. https://doi.org/10.3389/fimmu.2024.1403613 (2024).
Acknowledgement
This work was supported by the National Natural Science Foundation of China (Grant No. 81760442).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Y.X., W.X., Y.W., C.H. and Y.Z., the first draft of the manuscript was written by L.P., L.L., R.S. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, Y., Xiong, W., Wang, Y. et al. The immunosurveillance signature predicts the prognosis and immunotherapy sensitivity for colon adenocarcinoma. Sci Rep 15, 19899 (2025). https://doi.org/10.1038/s41598-025-03712-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-03712-2
