
Abstract
Despite notable advancements in prevention, medication, and treatment approaches, coronary artery disease (CAD) remains a significant challenge for healthcare systems and the economy. In relation to CAD, long non-coding RNAs (lncRNAs) can impact its development by influencing immune responses, affecting the functions of endothelial and vascular smooth muscle cells, and modifying lipid metabolism. Through the analysis of the GEO dataset (GSE42148), we identified differentially expressed genes (DEGs) and lncRNAs (DELs) in CAD patients. We performed functional enrichment and pathway analyses to clarify the roles of these DEGs. To investigate the interactions between DEGs and DELs, we created the lncRNA-mRNA interaction network. To investigate the interactions between DEGs and DELs, we constructed an lncRNA–mRNA interaction network. Candidate lncRNAs were validated by real-time PCR using peripheral blood from CAD patients. Our in vitro study confirmed that LINC00963 and SNHG15 were upregulated in CAD patients compared to the control group. Notably, LINC00963 levels were significantly elevated in patients with a positive family history, hyperlipidemia, hypertension, and diabetes, while SNHG15 expression was higher in smokers. Additionally, a significant negative correlation was found between the expressions of LINC00963 and SNHG15 and the age of the individuals. ROC curve analysis indicated that both lncRNAs have high sensitivity and specificity as biomarkers. Furthermore, this study suggests that LINC00963 and SNHG15 could serve as valuable markers for the early detection of CAD, particularly in younger individuals. It is proposed that these lncRNAs are associated with inflammatory conditions in CAD. Overall, LINC00963 and SNHG15 may act as promising early detection markers for CAD based on bioinformatics and peripheral blood-based validation.
Similar content being viewed by others

Introduction
Coronary artery disease (CAD) is a prevalent form of heart disease, accounting for approximately 2.2% of the global disease burden and 32.7% of all cardiovascular diseases. In 2020, around 19.05 million deaths worldwide were linked to cardiovascular diseases, reflecting an 18.71% increase since 20101. Survivors of myocardial infarction face a significantly elevated risk of subsequent events, with an annual mortality rate that is 5 to 6 times greater than that of individuals without CAD. CAD primarily affects the coronary arteries, which are the main blood vessels supplying blood to the heart. This condition leads to decreased blood flow to the heart muscle due to atherosclerosis, characterized by the accumulation of fats, cholesterol, and other substances on the artery walls, forming plaque that narrows the arteries. Key modifiable risk factors include hypertension, smoking, obesity, lipid levels, and psychosocial factors2,3.
Despite significant progress in prevention, medication, and treatment strategies, CAD continues to pose substantial healthcare and economic challenges. Therefore, there is a need for innovative diagnostic methods and new biomarkers for targeted therapies. Atherosclerosis is recognized as a chronic inflammatory ailment in which oxidative, hemodynamic, and biochemical factors disrupt endothelial cell function. This dysfunction results in alterations to endothelial permeability, the accumulation of macrophages, the release of inflammatory mediators, and the activation of smooth muscle cells, all of which further drive the progression of atherosclerosis4,5,6.
Long non-coding RNAs (lncRNAs) are a diverse group of non-coding RNA molecules that exceed 200 nucleotides in length and are characterized by their low conservation across species. Research suggests that lncRNAs play significant roles in various biological processes, including cell signaling, chromatin modification, and the regulation of transcription and translation, as well as their involvement in various human diseases4,7,8,9. In the context of CAD, lncRNAs can influence its pathogenesis by modulating immune responses, affecting the functions of endothelial cells and vascular smooth muscle cells, and altering lipid metabolism. Some lncRNAs may impact multiple pathways simultaneously7,10. A study by Shu He and colleagues identified PDXDC1-AS1 and SFI1-AS1 as sensitive and specific biomarkers for CAD, noting that they demonstrate synergistic effects when paired with certain environmental factors11.
Bioinformatics analysis has transformed the study of complex diseases by facilitating the identification of differentially expressed genes (DEGs), functional enrichment pathways, and molecular interaction networks. While bioinformatics is powerful for predicting regulatory networks, experimental validation is essential to confirm these predictions. This combined approach helps bridge the gap between in silico predictions and biological realities, providing a thorough understanding of the molecular mechanisms associated with diseases. In this study, this dual strategy for pinpointing reliable biomarkers is employed in the context of CAD research.
Materials and methods
Microarray data collection and differential expression analysis
The transcriptome profiles of mRNAs and lncRNAs were retrieved from the NCBI Gene Expression Omnibus (GEO) database using the dataset GSE42148 (https://www.ncbi.nlm.nih.gov/geo/)12. This dataset comprised 13 patients aged 40 to 55 years with angiographically confirmed CAD and 11 asymptomatic controls with normal ECG results. To detect differentially expressed genes (DEGs) and lncRNAs (DELs) between the CAD and control groups, the GEO2R web tool (http://www.ncbi.nlm.nih.gov/geo/geo2r/) was employed (accessed March 2024; default Benjamini–Hochberg correction used). Genes were considered differentially expressed based on a |log2 fold change (log2FC)| ≥ 1 and a p-value < 0.05.
Functional enrichment analysis
To investigate the underlying biological mechanisms, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed on the identified DEGs using the Database for Annotation, Visualization, and Integrated Discovery (DAVID v2023q4) (https://david.ncifcrf.gov)13. A p-value < 0.05 was set as the significance threshold for all analyses. The GO analysis focused on three main categories: molecular function (MF), cellular component (CC), and biological process (BP). Statistically significant pathways were selected based on their enrichment scores and associated p-values.
Construction of the lncRNA-mRNA network
The lncRNA-mRNA interaction network was constructed using Cytoscape to explore the functional interactions among DEGs and DEL. Each lncRNA was analyzed using the StarBase platform(https://rnasysu.com/encori/index.php) to find mRNA targets14.
Study population
This study was conducted following approval from the Ethics Committee of Hormozgan University of Medical Sciences (approval code: “IR.HUMS.REC.1403.303”), and the study protocols were performed in accordance with the relevant guidelines and regulations. Also, the Declaration of Helsinki was followed15. The legal guardians of all participants provided written informed consent. Blood samples were taken in a fasting state from 50 patients diagnosed with CAD and 50 healthy individuals. Participants were recruited from Seyed al-Shohada Heart Hospital, affiliated with Urmia University of Medical Sciences.
RNA extraction and cDNA synthesis
A total of 10 mL of blood was drawn from each participant using EDTA-coated tubes to inhibit coagulation. Following the manufacturer’s protocol, total RNA was extracted from the blood samples of individuals with CAD and healthy controls using the RNX Plus™ kit (Cat. no: RN7713c/EX6101; Sinaclon, Iran). To eliminate any DNA contamination, RNA samples were treated with RNase-free DNase (Qiagen, Valencia, CA, USA). The RNA’s quality and integrity were evaluated using a Thermo Scientific™ NanoDrop 2000 spectrophotometer and further validated via DNase I treatment (Cat. no: EN0521; Thermo Fisher Scientific, USA) and 2% agarose gel electrophoresis. Following this, 2.0 µg of RNA from each sample underwent complementary DNA (cDNA) synthesis using the Yektatajhiz cDNA Synthesis Kit (Cat. no: YTA4500; Yektatajhiz, Iran) in a 20 µL reaction.
Quantitative Real-Time PCR
Quantitative Real-Time PCR (qRT-PCR) was carried out in 10 µl volumes (in triplicate) using the Mic qPCR system and SYBR Green master mix (Yektatajhiz, Iran) The SNHG15 and LINC00963 gene expression levels were analyzed, with SRSF4 as the internal reference gene. The sequences of primers used for these genes are detailed in Table 1.
Statistical analysis
All data are expressed as mean ± standard deviation (SD), obtained from a minimum of three independent replicates. Statistical evaluations were performed using SPSS software version 27.0 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism version 8.0 (GraphPad Software, San Diego, USA). The Mann-Whitney U test was utilized for group comparisons, with results deemed statistically significant at P < 0.05. Spearman’s correlation coefficient was also applied to the correlation analysis.
Data visualization
The analysis findings were visualized using the SRplot platform (https://bioinformatics.com./srplot), which facilitated the creation of various plot types, including volcano plots, chord plots, bubble plots, enriched horizontal bar plots (accessed April 2024; latest version)16. In addition, Cytoscape (v3.10.1) (https://cytoscape.org/) was employed to analyze and visualize the regulatory subnetwork, hub genes, and the lncRNA-mRNA network17. Furthermore, the ggplot2 R package was used to generate volcano plots, enhancing the graphical representation of DEGs18.
Results
Microarray data analysis and differential expression analysis
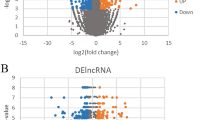
The differential expression analysis of mRNAs and lncRNAs between CAD patients and controls revealed significant changes in gene expression. Using the GEO2R tool on the GSE42148 dataset, 62,972 genes were analyzed, of which 171 were significantly down-regulated and 458 were up-regulated. These genes were identified based on the criteria of |log2FC| ≥ 1 and p-value < 0.05 (Fig. 1).
322 protein-coding genes and 25 lncRNAs were differentially expressed in CAD patients compared to healthy controls (supplementary Table 1). The results are visually represented in the Volcano Plot, where up-regulated genes are shown in red, down-regulated genes in blue, and non-significant genes in grey. This analysis highlights the transcriptional changes underlying CAD pathogenesis.

Volcano plot showing differentially expressed genes between CAD patients and healthy controls. Differential gene expression analysis was conducted on the GSE42148 dataset using GEO2R (up-regulated genes are shown in red, down-regulated genes in blue, and non-significant genes in grey).
Functional enrichment analysis
GO enrichment analysis was conducted to explore the biological functions of DEGs linked to CAD. This examination uncovered significant enrichment in BPs, MFs, and CCs, providing insights into the molecular and cellular mechanisms involved in CAD.
The key BPs identified included the enhanced regulation of transcription by RNA polymerase II, the inflammatory response, signal transduction, and intercellular signaling. Additionally, other significant processes included the upregulation of tumor necrosis factor production, T cell proliferation during the immune response, axon extension, and the downregulation of cell growth and transcription. In the category of MFs, enriched terms highlighted the roles of protein binding, DNA-binding transcription activator activity, peptide hormone binding, and CXCR chemokine receptor binding. These MFs emphasize the significance of transcription regulation and receptor-ligand interactions in CAD. The CCs associated with the DEGs included the cytoplasm, cytosol, intracellular membrane-bounded organelles, specific granule membranes, and tertiary granule membranes. Additional enrichment in components such as the extracellular space and ficolin-1-rich granule membranes reflects the compartmentalized activities underlying CAD pathology (Fig. 2 A).
To further explore the involvement of DEGs in broader biological pathways, KEGG pathway enrichment analysis was performed19,20,21. This analysis identified several key pathways significantly associated with CAD, including the IL-17 signaling pathway, Ras signaling pathway, and pathways in cancer (Fig. 2B).

Functional enrichment analysis. (A) GO enrichment analysis revealed significant enrichment in BPs, MFs, and CCs, shedding light on the molecular and cellular mechanisms involved in CAD. (B) KEGG pathway enrichment analysis identified several key pathways significantly associated with CAD.
Construction of the lncRNA-mRNA network
The lncRNA-mRNA interaction network was constructed using Cytoscape to explore the functional interactions among DEGs and DEL. Each lncRNA was analyzed using the StarBase platform to find mRNA targets. Of 25 DEL, 17 had interactions with mRNA targets. The regulatory interactions between lncRNAs and mRNAs of DEGs were further analyzed through the construction of the lncRNA-mRNA network. This analysis identified 8 key lncRNAs (including LINC00963, PVT1, GABPB1-AS1, MIR34AHG, MIR4435-2HG, ZNF667-AS1, ST20-AS1, and SNHG15) that target genes and regulate their expression. Among the 8 key lncRNAs, LINC00963 and SNHG15 were selected for validation based on their network centrality in the lncRNA–mRNA regulatory network, novelty, and our previous research on these lncRNAs on inflammatory diseases like rheumatoid arthritis (RA) and Systemic lupus erythematosus (SLE). The lncRNA-mRNA network provides a visual representation of these interactions and highlights the role of non-coding RNAs in the regulation of gene expression in CAD (Fig. 3).

lncRNA–mRNA interaction network constructed from DELs and their predicted mRNA targets. Nodes represent genes: red for lncRNAs and blue for mRNAs. Node size corresponds to degree centrality.
The expression of LINC00963 and SNHG15 in patients
The qRT-PCR validated LINC00963 and SNHG15 expression in patients and the control group. The results showed that both LINC00963 and SNHG15 expressions were upregulated in CAD patients compared to normal samples. In CAD samples, LINC00963 and SNHG15 expressions were 5.91-fold (P < 0.0001) and 2.30-fold (P < 0.0001) compared with the control group, respectively (Fig. 4).
Additional analysis revealed that LINC00963 expression in patients with hyperlipidemia, hypertension, and diabetes was significantly (P < 0.05) higher than in patients with normal conditions for these parameters (Fig. 5A-C). Also, it was found that patients with a previous family history of CAD had a significantly (P < 0.05) higher expression of LINC00963 (Fig. 5E). Still, there was no differentiation between smoker and non-smoker patients (Fig. 5D).
On the other hand, the SNHG15 expression doesn’t show significant differentiation in patients with hyperlipidemia, hypertension, diabetes, and positive family history for CAD compared with patients with normal conditions for these parameters (Fig. 6A-E). Still, it was found that a smoker patient had a significantly (P < 0.05) higher expression of SNHG15 (Fig. 6D).

SNHG15 and LINC00963 expression were validated in CAD patients versus healthy controls using qRT-PCR. (A) qRT-PCR analysis revealed a significant upregulation of SNHG15 expression in CAD patients (n = 50) compared to healthy controls (n = 50) (P < 0.001; Mann–Whitney U test). (B) Heatmap visualization of SNHG15 expression across individual samples, showing consistent elevation in the CAD group. (C) LINC00963 expression was also significantly higher in CAD patients than in controls (P < 0.001). (D) Heatmap of LINC00963 expression confirms the strong upregulation pattern in CAD individuals. Data are presented as mean ± SD. *** indicates statistical significance (P < 0.001). All reactions were performed in biological duplicates and technical triplicates. These results support the potential of SNHG15 and LINC00963 as diagnostic biomarkers in CAD.
LINC00963 and SNHG15 correlations with age
The correlation analysis was done to recognize the association of LINC00963 and SNHG15 expressions with the age of patients. The results showed that LINC00963 expression has a negative correlation with age in CAD patients (r = 0.322, p = 0.03) but not in the control group (Fig. 7A, B). Also, there were negative correlations between age and SNHG15 expression in CAD (r = 0.684, p < 0.0001) and control groups (r = 0.827, p < 0.0001) (Figs. 7 C, D).

SNHG15 and LINC00963 expression were validated in CAD patients versus healthy controls using qRT-PCR. (A) qRT-PCR analysis revealed a significant upregulation of SNHG15 expression in CAD patients (n = 50) compared to healthy controls (n = 50) (P < 0.001; Mann–Whitney U test). (B) Heatmap visualization of SNHG15 expression across individual samples, showing consistent elevation in the CAD group. (C) LINC00963 expression was also significantly higher in CAD patients than in controls (P < 0.001). (D) Heatmap of LINC00963 expression confirms the strong upregulation pattern in CAD individuals. Data are presented as mean ± SD. *** indicates statistical significance (P < 0.001). All reactions were performed in biological duplicates and technical triplicates. These results support the potential of SNHG15 and LINC00963 as diagnostic biomarkers in CAD.

Analysis of SNHG15 expression in CAD patients with different clinical characteristics. Box plots represent the relative expression levels of SNHG15 in subgroups of CAD patients categorized by clinical variables: (A) No significant difference was observed in SNHG15 expression between patients with and without hyperlipidemia (ns). (B) SNHG15 levels did not significantly differ between hypertensive and normotensive individuals (ns). (C) Diabetic and non-diabetic CAD patients showed comparable expression levels of SNHG15 (ns). (D) A significantly higher expression of SNHG15 was detected in smokers compared to non-smokers (P < 0.001). (E) No significant association was found between SNHG15 expression and CAD (ns) family history. Statistical analyses were performed using the Mann–Whitney U test. *** = P < 0.001; ns = not significant.

Correlation analysis between age and lncRNA expression in CAD and control groups. Spearman correlation was performed to assess the relationship between patient age and the expression levels of LINC00963 and SNHG15: (A) A modest but statistically significant negative correlation was observed between LINC00963 expression and age in CAD patients (r = − 0.3225, P = 0.0223). (B) No significant correlation was found between LINC00963 expression and age in the control group (r = − 0.0616, P = 0.6711). (C) A strong negative correlation was detected between SNHG15 expression and age in CAD patients (r = − 0.6845, P < 0.0001). (D) In the control group, SNHG15 expression showed a very strong inverse correlation with age (r = − 0.8271, P < 0.0001). Red lines indicate linear regression fit.
ROC curve analysis
The ROC curve analysis was performed to determine the sensitivity and specificity of the LINC00963 and SNHG15 expressions as biomarkers. The Area under the ROC curve (AUC) was 0.94 (p < 0.0001) for LINC00963 in CAD patients. Also, the SNHG15 received a 0.81 (p < 0.0001) AUC (Fig. 8).

Receiver Operating Characteristic (ROC) curve analysis evaluating the diagnostic performance of LINC00963 and SNHG15 in distinguishing CAD patients from healthy controls. (A) ROC analysis of LINC00963 expression yielded an area under the curve (AUC) of 0.9424 (95% CI: 0.8981–0.9867, P < 0.0001), indicating high diagnostic accuracy. (B) SNHG15 showed an AUC of 0.8196 (95% CI: 0.7374–0.9018, P < 0.0001), supporting its moderate diagnostic value.
Discussion
Overall, our in silico analyses highlighted 322 protein-coding and 25 lncRNAs that show significant differentiation in CAD. The results of functional enrichment analysis showed that components of the immune system and the inflammatory process, transcriptional regulators, and receptor-ligand interactions are the most important biological roles of DEGs identified in CAD. lncRNA-mRNA network revealed 8 lncRNAs (LINC00963, PVT1, GABPB1-AS1, MIR34AHG, MIR4435-2HG, ZNF667-AS1, ST20-AS1, and SNHG15) that related to the DEGs.
Our in vitro investigation approved the upregulation of LINC00963 and SNHG15 in CAD patients in comparison with the control group. Also, it was revealed that LINC00963 expression in patients with positive family history, hyperlipidemia, hypertension, and diabetes was significantly higher compared with others. Also, SNHG15 expression was significantly higher in smoker patients. There was a significant negative correlation between LINC00963 and SNHG15 expression and the age of individuals.
Atherosclerosis is a chronic inflammatory condition in which the immune system plays a vital role in its initiation and development. This inflammation causes endothelial dysfunction, increasing its permeability to lipoproteins and leading to their accumulation beneath the endothelial layer22. Additionally, this process promotes the recruitment of white blood cells and the activation of platelets. Monocytes that migrate to the affected area differentiate into macrophages, which can take on either pro-inflammatory or anti-inflammatory traits depending on their microenvironment23. The dynamics between these macrophage types influence the progression or resolution of atheroma.
Both macrophages and smooth muscle cells produce inflammatory cytokines such as IL-1β, IL-12, and IL-6. Within the arterial walls, low-density lipoprotein (LDL) cholesterol undergoes oxidation, while triglyceride-rich and remnant lipoproteins further drive inflammation. Macrophages actively break down these oxidized lipoproteins, leading to the formation of a lipid-rich necrotic core encased by a collagen fibrous cap, ultimately resulting in the development of fibro-atheromas24. In the context of chronic inflammation, macrophages may degrade this fibrous cap, creating a thin-cap fibro-atheroma, which increases the risk of plaque rupture23,24.
The long intergenic noncoding RNA 00963 (LINC00963) is located on chromosome 9q34.11 in humans. Its abnormal expression has previously been associated with cancer progression by affecting various cellular processes such as proliferation, migration, invasion, epithelial-mesenchymal transition (EMT), and apoptosis25. Elevated levels of LINC00963 are associated with different cancer clinicopathological features and worse patient outcomes, suggesting its potential as a biomarker for diagnosing hepatocellular carcinoma26. LINC00963 can create a complex competing endogenous RNA (ceRNA) network by interacting with 22 distinct miRNAs across 14 cancer types. It also has a direct impact on four protein-coding genes25. Specifically, LINC00963 promotes the transition of prostate cancer from an androgen-dependent state to an androgen-independent state by participating in the transactivation of EGFR27. Furthermore, LINC00963 interacts with EZH2, which reduces p21 expression, thus promoting the proliferation and invasion of glioma cells. In non-small cell lung cancer, LINC00963 brings together NONO and CRTC, generating a positive feedback loop (LINC00963/NONO/CRTC/CREB/LINC00963) that enhances cancer cell metastasis28.
On the other hand, lncRNA SNHG15 has emerged as a significant player in the onset and progression of various human cancers due to its dysregulation within malignant tumors29. SNHG15 is often overexpressed in various cancers, including lung, gastric, colorectal, and liver cancers, and its levels correlate with disease severity and poor prognosis30,31,32,33. This lncRNA influences critical processes such as cell proliferation, apoptosis, metastasis, and nutrient evasion in response to cancer therapy. SNHG15 regulates various oncogenes and tumor suppressor genes through molecular mechanisms involving microRNA sponging30,31,32,33.
Based on the role of LINC00963 and SNHG15 in various cancers linked to inflammation, along with the findings of this study, it can be inferred that this lncRNA is closely associated with inflammatory conditions. The ROC curve analysis demonstrated that LINC00963 and SNHG15 exhibit high sensitivity and specificity as biomarkers. Additionally, the current study suggests these two lncRNAs could serve as promising markers for the early detection of CAD in younger individuals. Further research involving larger populations and a deeper investigation into the functions of these lncRNAs will enhance our understanding of their roles.
Although our study successfully identified and validated LINC00963 and SNHG15 as potential biomarkers for CAD, we acknowledge the absence of functional validation experiments. Future studies are warranted to investigate their mechanistic roles in vascular cells, such as endothelial or smooth muscle cells, through knockdown or overexpression models to assess their effects on inflammation, migration, and proliferation.
Conclusion
It was revealed that LINC00963 and SNHG15 gene expression are significantly upregulated in CAD patients. It was suggested that these lncRNAs are linked to the inflammatory condition in CAD. Also, it was suggested that LINC00963 and SNHG15 could serve as promising markers for the early detection of CAD in younger individuals. While our findings highlight LINC00963 and SNHG15 as promising diagnostic biomarkers for CAD, further functional studies are necessary to elucidate their precise roles in CAD pathophysiology.
Data availability
The sequence data supporting the findings of this study are derived from publicly available dataset: GSE42148. These datasets are accessible through the Gene Expression Omnibus (GEO) repository https://www.ncbi.nlm.nih.gov/geo/. The experimental data that support the findings of this study are available from the corresponding author (Pegeah Mousavi), upon reasonable request.
References
Tsao, C. W. et al. Heart disease and stroke Statistics-2023 update: A report from the American heart association. Circulation 147 (8), e93–e621 (2023).
Nakahara, T. et al. Coronary artery calcification: from mechanism to molecular imaging. JACC Cardiovasc. Imaging. 10 (5), 582–593 (2017).
Shahjehan, R. D., Sharma, S. & Bhutta, B. S. Coronary Artery Disease, in StatPearls [Internet] (StatPearls Publishing, 2024).
Matsuzawa, Y. and A.J.C.a.d. Lerman, Endothelial dysfunction and coronary artery disease: assessment, prognosis, and treatment. Coronary artery disease. 25(8): pp. 713–724. (2014).
Ding, L. et al. Gene expressions underlying mishandled calcium clearance and elevated generation of reactive oxygen species in the coronary artery smooth muscle cells of chronic heart failure rats. Chinese Medical Journal. 130(04): pp. 460–469. (2017).
Nurnberg, S. T. et al. Coronary artery disease associated transcription factor TCF21 regulates smooth muscle precursor cells that contribute to the fibrous cap. PLoS genetics. 11(5): p. e1005155. (2015).
Ghafouri-Fard, S., Gholipour, M. & Taheri, M. The Emerging Role of Long Non-coding RNAs and Circular RNAs in Coronary Artery Disease. 8. (2021).
Nishikawa, K. J.B.r. Kinjo, Essential role of long non-coding RNAs. De Novo Chromatin Modifications: Genomic Address Code Hypothesis. 9, 73–77 (2017).
Peng, W. X., Koirala, P. & Mo, Y. Y. J. O. LncRNA-mediated Regul. Cell. Signal. cancer 36(41): 5661–5667. (2017).
Li, L. et al. Characterization of LncRNA expression profile and identification of novel LncRNA biomarkers to diagnose coronary artery disease. Atherosclerosis 275, 359–367 (2018).
He, S. et al. Long non-coding RNA in Coronary Artery Disease: the Role of PDXDC1-AS1 and SFI1-AS123p. 219 (Functional & Integrative Genomics, 2023). 3.
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res, 30(1): pp. 207 – 10. (2002).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50 (W1), W216–w221 (2022).
Li, J. H. et al. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42 (D1), D92–D97 (2013).
Shrestha, B. & Dunn, L. The declaration of Helsinki on medical research involving human subjects: a review of seventh revision. (2019).
Tang, D. et al. SRplot: A free online platform for data visualization and graphing. PLoS One. 18 (11), e0294236 (2023).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504 (2003).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-, 2016).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–d677 (2025).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Libby, P. The changing landscape of atherosclerosis. Nature 592 (7855), 524–533 (2021).
Henein, M. Y. et al. The role of inflammation in cardiovascular disease. Int. J. Mol. Sci. 23 (21), 12906 (2022).
Libby, P. & Hansson, G. K. From focal lipid storage to systemic inflammation: JACC review topic of the week. J. Am. Coll. Cardiol. 74 (12), 1594–1607 (2019).
Xie, Z. et al. LINC00963: A potential cancer diagnostic and therapeutic target. Biomed. Pharmacother. 150, 113019 (2022).
Wu, J. H. et al. LINC00963 Promotes Hepatocellular Carcinoma Progression by Activating PI3K/AKT Pathway22 (European Review for Medical & Pharmacological Sciences, 2018). 6.
Sun, F. et al. Long noncoding RNA LINC00963 induces NOP2 expression by sponging tumor suppressor miR-542-3p to promote metastasis in prostate cancer. Aging (Albany NY). 12 (12), 11500 (2020).
Ye, F. et al. LINC00963 confers oncogenic properties in glioma by regulating the miR-506/BCAT1 Axis. Cancer Manage Res. 12, 2339–2351 (2020).
Zhang, N. et al. Long noncoding RNA SNHG15: A promising target in human cancers. Front. Oncol. 13, 1108564 (2023).
Saeinasab, M. et al. SNHG15 is a bifunctional MYC-regulated noncoding locus encoding a LncRNA that promotes cell proliferation, invasion and drug resistance in colorectal cancer by interacting with AIF. J. Experimental Clin. Cancer Res. 38, 1–16 (2019).
Huang, J. et al. LncRNA SNHG15 regulates EGFR-TKI acquired resistance in lung adenocarcinoma through sponging miR-451 to upregulate MDR-1. Cell Death Dis. 11 (7), 525 (2020).
Chen, Z. et al. LncRNA SNHG15 modulates gastric cancer tumorigenesis by impairing miR-506-5p expression. Biosci. Rep. 41 (7), BSR20204177 (2021).
Ye, J. et al. LncRNA SNHG15 promotes hepatocellular carcinoma progression by sponging miR-141‐3. J. Cell. Biochem. 120 (12), 19775–19783 (2019).
Acknowledgements
We express our sincere gratitude for the technical help provided by the Vice-Chancellor for Research at Hormozgan University of Medical Sciences. Hormozgan University of Medical Sciences provided financial assistance for this study (grant no: 4030313).
Funding
The Hormozgan University of Medical Sciences provided financial assistance for this study (grant nu: 4030313).
Author information
Authors and Affiliations
Contributions
MS and PN conceived and designed the study. OK and NJ performed clinical examinations and acquired clinical samples. MS and VSR performed the q-PCR verification experiment. MS and AG conducted the data acquisition and analysis and drafted and wrote the manuscript. PM and NJ supervised, managed the data, and reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript and accept personal responsibility for the author’s contribution.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was conducted following approval from the Ethics Committee of Hormozgan University of Medical Sciences (approval code: “IR.HUMS.REC.1403.303”), and the study protocols were performed in accordance with the relevant guidelines and regulations. Also, the Declaration of Helsinki was followed15.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saberiyan, M., Noorabadi, P., Kahourian, O. et al. Integrative in Silico and in vitro validation suggest LINC00963 and SNHG15 as candidate biomarkers for coronary artery disease. Sci Rep 15, 21501 (2025). https://doi.org/10.1038/s41598-025-08777-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-08777-7
