
Abstract
Based on single-stranded DNA library method, we established an efficient workflow to parallelly construct targeted genomic and epigenomic sequencing libraries from a small amount of DNA. We applied the protocol to nine pediatric brain cancer DNA samples containing various extents of damage from formalin fixation and/or DNA oxidation. Compared to our previous study, the new exome protocol showed superior uniformity of coverage. Many artifactual mutation calls introduced by DNA damages were eliminated by bioinformatics filtering tools. After filtration, 89.4–97.0% of somatic single nucleotide variant (SNV) calls generated by double-stranded DNA library were reproduced in formalin-fixed paraffin-embedded (FFPE) samples, which was achieved with substantially reduced DNA input amounts (26.7-50ng). In methylome analysis, we obtained methylation calls for 78–92% of target CpGs with at least 10x coverage when using 100ng of FFPE DNA, which is comparable to those obtained from fresh frozen samples. We also obtained SNV calls from methylome data, recovering 39–76% of filtered SNVs from exome data in nine brain cancer samples. In conclusion, we present a simple protocol for parallel construction of targeted exome and methylome sequencing libraries, which was successfully applied to damaged brain cancer DNA samples from FFPE tissues stored for prolonged periods.
Similar content being viewed by others

Introduction
Next-generation sequencing (NGS) enables the collection of vast amounts of information from various clinical resources. This powerful tool also facilitates the investigation of multiple aspects from a single sample1,2. In a recent study, NGS-based combined genomic and epigenomic signatures in plasma sample demonstrated an enhanced sensitivity for detection of minimal residual disease in colorectal cancer patients compared to use of genomic signatures alone3. Simultaneous acquisition of genomic and epigenomic alterations from a single sample is invaluable in both clinical and research aspects. Most NGS-based studies require fresh-frozen material due to the need for high-quality of DNA. However, formalin-fixed paraffin-embedded (FFPE) tissue represents one of the major sources of biospecimens for molecular analysis, especially in clinical settings due to its cost-effective long-term storage capabilities. However, it is well known that a wide range of damage occurs including DNA fragmentation and nucleic acid-protein crosslinking during formalin fixation process and/or in improper storage conditions4, which makes it challenging to obtain a high-quality NGS data from FFPE samples. In addition, DNA methylation profiling requires bisulfite treatment to convert unmethylated cytosine to uracil to distinguish unmethylated from C5-methylated cytosines. The bisulfite conversion process causes additional DNA degradation, which raises further challenges for DNA methylation profiling with DNA from FFPE5.
The main purpose of this study is to establish an efficient preparation method for exome and methylome sequencing libraries that can be applied to a small amount of damaged DNA sample such as DNA from FFPE tissues. In this study, we provide a simple and efficient method for parallel construction of target-enriched exome and methylome NGS libraries using single strand-based library preparation method, which allows utilization of DNA fragments containing nicks as well as single-stranded DNA molecules. The protocol enabled us to obtain reliable targeted exome and methylome NGS data with relatively low amounts of DNA in nine pediatric brain cancer DNA samples containing various extents of DNA damage.
Materials and methods
Reference DNA materials and pediatric brain cancer DNA samples
The following reference genomic DNA (gDNA) materials were commercially sourced: NA12878 gDNA (Coriell Cell Repositories), EpiScope Methylated HCT116 gDNA (Takara Bio Inc.), and EpiScope Unmethylated HCT116 DKO gDNA (Takara Bio Inc.). Reference gDNAs with various methylation proportions (100%, 70%, 30%, and 0% methylated) were prepared by mixing Methylated and Unmethylated HCT116 gDNA. For clinical samples, we used total nine gDNA samples of pediatric brain cancer, a subset of those from our previous study, including eight medulloblastoma and one secondary glioblastoma sample6. This study was approved by the institutional review board (IRB) of Jeonbuk National University (IRB JBNU-2023-05-025).
Preparation of targeted exome and methylome sequencing libraries
DNA was sheared to a size range of 150–250 bp using focused-ultrasonicator (Covaris, USA) and final yield and quality were assessed using the Agilent 4150 TapeStation (Agilent Technologies, Germany). Absorbance was measured using a Qubit (Thermo Fisher Scientific, Wilmington, USA). For methylome library, bisulfite conversion was performed with 100ng of sheared DNA using the EZ DNA Methylation-Lightning Kit (Zymo Research, USA) according to the manufacturer’s instructions. The concentration of bisulfite-treated DNA was measured using the Qubit ssDNA Assay Kit (Thermo Fisher Scientific, USA). Subsequently, samples were denatured to single-stranded DNA with single strand-based SRSLY NanoPlus DNA NGS Library Preparation Base kit (Claret Bioscience, USA) followed by adapter ligation and indexing PCR with SRSLY UDI Primer Set (Claret Bioscience, USA). In the indexing PCR step, Q5U Master Mix (NEB, USA) was used for methylome library preparation with bisulfite converted DNA. Library fragment size was measured using a D1000 ScreenTape and Reagents Kit (Agilent Technologies, Germany), and the yields were quantified by Qubit (Thermo Fisher Scientific, USA). Target enrichment was performed using Twist Exome 2.0 Panel (Twist Bioscience, USA) and Twist Human Methylome Panel (Twist Bioscience, USA), respectively. Library fragment size was measured using a D1000 ScreenTape and Reagents Kit (Agilent Technologies, Germany), and the yields were quantified by Qubit (Thermo Fisher Scientific, USA). The final constructed libraries were sequenced by PE150 strategy on the Illumina NovaSeq6000 platform at Macrogen Inc. (Macrogen, Korea). The generated exome and methylome datasets were deposited into sequence read archive (SRA) database under accession number PRJNA1144056.
Next generation sequencing data analyses
For exome data analysis, raw reads were trimmed with TrimGalore (v0.6.10) (--2colour 20). Trimmed reads were aligned to the GRCh38 reference genome with Burrows-Wheeler aligner (BWA)-MEM (v0.7.17). Mapped reads were deduplicated with Picard MarkDuplicates (v2.27.4) and base recalibration was applied with BaseRecalibrator and ApplyBQSR tools of Genome Analysis ToolKit (GATK) (v4.5.0). For nine brain cancer samples, somatic variants were called with GATK Mutect2 in tumor-only mode with an option of -pon using downloaded variant call format (VCF) dataset of public panel of normals (https://storage.cloud.google.com/gatk-best-practices/somatic-hg38/1000g_pon.hg38.vcf.gz). Further filtering steps were performed with MicroSEC7, FFPolish8, Strand Orientation Bias Detector (SOBDetector)9, and/or GATK LearnReadOrientationModel followed by GATK Mutect2 FilterMutectCalls. All the analyses were repeated with the exome sequencing data from our previous study (PRJNA418669 in the SRA database)6. For the analysis of exome data from a reference NA12878 gDNA sample, GATK HaplotypeCaller was applied to the base recalibrated BAM file and the resulting VCF was subject to variant filtration using CNNScoreVariants with 1D and 2D models.
For the analysis of methylome data, raw reads were trimmed with TrimGalore (--2colour 20, --clip_R1 8, --three_prime_clip_R1 8, --clip_R2 8, --three_prime_clip_R2 8). Trimmed reads were mapped to GRCh38 and deduplicated with Bismark (v0.24.2)10. Subsequently, strand-specific or destranded methylation calls were extracted using Bismark bismark_methylation_extractor or coverage2cytosine module, respectively. Finally, we used recently developed BISulfite-seq Command line User Interface Toolkit (BISCUIT) to call SNVs from methylome data: the trimmed reads were aligned to GRCh38 and pileup VCF files were generated using BISCUIT11.
A metric to assess coverage uniformity in sequencing data, fold-80 base penalty, was calculated with CollectHsMetrics of GATK12. The fold 80 base penalty is a metric for smoothness of coverage, indicating the fold-over coverage needed to bring 80% of target bases to the mean coverage level. A value of 1 is theoretically perfect, while a value of 2 suggests repeating sequencing to achieve satisfactory coverage. In general, a fold 80 base penalty of 3 or less is considered satisfactory, a well-balanced coverage.
Public datasets for NA12878
Two public VCF files of NA12878, HG00113 and GSE20625314, were downloaded from https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/NISTv4.2.1/GRCh38/HG001_GRCh38_1_22_v4.2.1_benchmark.vcf.gz and the gene expression omnibus (GEO) database, respectively. Three public CpG methylation call datasets of NA12878, GSM277252415, GSM634830516, and GSM634830616, were downloaded from the GEO database. The genomic coordinates of the methylation call of GSM2772524 were converted from hg19 to GRCh38.
Results
Establishment of a fast and simple protocol for parallel preparation of exome and methylome sequencing libraries with single strand-based method
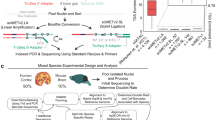
The schematic diagram of the established protocol is presented in Fig. 1, starting with 26.7-50ng and 33.4-100ng of sheared DNA for exome and methylome libraries, respectively (Table 1). The process involves shearing of DNA with a length of 150–250 bp, bisulfite conversion for methylome, denaturation, followed by adapter ligation and cleanup, and 10 cycles of PCR amplification and cleanup. For subsequent target enrichment, the prepped SRSLY library is hybridized with Twist Human Exome v2.0 Panel or Human Methylome Panel. Except for the bisulfite conversion step, all steps were carried out parallelly, which takes approximately 1.5 days to perform including overnight hybridization process. If the overnight hybridization step is replaced by a fast one which takes 3 to 4 h, the whole process can be performed within 1 day. In contrast to the most conventional methods that typically require distinct reagents and time-consuming protocols for each library construction, we used unified protocols and reagents for both exome and methylome libraries, which eventually improved efficiency and cost-effectiveness in library construction.

Overall workflow of parallel construction of targeted exome and methylome sequencing libraries.
Quality assessment of investigated DNA samples and prepped sequencing libraries
To assess the performance of the established protocol, we included nine pediatric brain cancer DNA samples from our previous study6 and five standard reference gDNA materials (Table 1). The pediatric brain cancer samples included four DNA samples from FFPE tissues, designated as P1 to P4, and five from fresh frozen tissues, designated as F1 to F5. The four FFPE tissues had been stored for 1 to 10 years before the DNA was extracted, while the five fresh frozen tissues had been cryopreserved for several months to 12 years. All brain cancer DNA samples were extracted approximately 5 years prior to the NGS library construction in current study. Additionally, a reference NA12878 gDNA was used for construction of exome and methylome libraries. For an additional reference analysis of methylome, four control gDNA mixtures with varying methylation proportions, 0, 30, 70, and 100%, were generated by mixing Methylated HCT116 gDNA (highly methylated) and HCT116 DKO gDNA (< 5% of CpG-methylation).
In standard gel electrophoresis, considerable degradation was noticed in three FFPE DNA samples (P1 – P3) (Figure S1). All sequencing libraries were constructed successfully (Figure S2) even in one brain cancer sample with limited DNA availability (P2), for which the libraries were produced using 26.7ng and 33.4ng of DNA for exome and methylome, respectively (Table 1). Detailed information of the sequencing libraries is provided in Table S1.
Evaluation of performance of exome sequencing
Basic statistics of exome sequencing data from current and previous study are summarized in Table S2. In nine brain cancer samples, current sequencing data were generated with mean target coverages of 36.3-66.2x, along with duplication rates of 20.2–38.2%. In contrast, the result of previous study showed much higher mean target coverages of 109.9-167.4x and lower duplication rates of 2.6–27.5%. The higher duplication rates of the current study can be explained by the crucial difference in the amount of input DNA: a much lower amount of DNA (26.7 or 50ng) was used in the current study, whereas a significantly higher amount (500-1,000ng) was used in the previous study. Notably, off-target rates of the current study ranged from 10.3 to 17.3%, which was considerably lower compared to those of the previous study, 18.7–37.6%.
On the other hand, superior uniformity of coverage was evidenced by more evenly distributed depth of coverages (Fig. 2a) and lower levels of fold 80 base penalty (Wilcoxon rank sum test p-value: 0.0012) (Fig. 2b), suggesting that our exome protocol performed better with relatively lower depths.

Exome sequencing statistics and detection of artifactual base changes in somatic single nucleotide variant (SNV) calls in nine pediatric brain caner samples in current (Twist) and previous (Agilent) studies. (a) Distribution of depth of coverage. (b) Estimated fold 80 base penalties. (c) Median insert size calculated from uniquely aligned properly paired reads (mapping quality ≥ 20). (d) Distribution of insert size of uniquely aligned properly paired reads (mapping quality ≥ 20). (e) Number of primarily-called and SOBDetector-filtered autosomal SNV calls (dpeth ≥ 10). (f) Number of primarily-called and filtered autosomal SNV calls in context of SNV type (dpeth ≥ 10).
The median and distribution of insert size were considerably shorter in the current study (Wilcoxon rank sum test p-value: 0.0019) (Fig. 2c and d). In general, compared to double strand-based DNA libraries, single strand-based ones are more enriched with short DNA capturing ultrashort double-stranded DNA and single-stranded DNA and double-stranded DNA with nicks17,18. Several sequencing strategies were also disparate between the two studies including exome capture kits (Twist Human Exome v2.0 Panel vs. Agilent SureSelect Human All Exon V5), sequencing read lengths (2 ⨯ 151 vs. 2 ⨯ 101 bp), and sequencing platforms (Illumina NovaSeq 6000 vs. HiSeq 2000 or 2500) (Table S2).
We firstly compared somatic single nucleotide variant (SNV) calls detected in current and previous studies in nine brain cancer samples. Since the two studies captured nonidentical target regions, we investigated only the common target region (~ 31 Mb). In addition, we employed a well-known computational filtering tool, SOBDetector9, to remove artifactual SNV calls that might be introduced during formalin fixation, long-term storage, or DNA shearing process19,20,21. Figure 2e shows the total number of SNVs that were primarily called (depth ≥ 10) and the number of SNVs that retained after excluding artifacts by the SOBDetector9. A remarkable number of initially called variants were filtered out, which was most prominent in three FFPE samples of current study (Twist P1 – P3) (Fig. 2e left panel). When considering variant type of SNVs, the removed artifactual calls were mainly confined to two types of base changes, C > T/G > A and C > A/G > T (Fig. 2f). It has been known that the artifactual C > T/G > A base changes are introduced by hydrolytic deamination of cytosine residues by formalin fixation process, which increases in proportion to the storage duration of FFPE tissues20,21. As expected, the highest proportion of artifactual removal of C > T/G > A calls (48.8%) was observed in a Twist FFPE sample, P1 (Table S3), for which the DNA was extracted after a 10-year storage duration (Table 1). Approximately 35% of C > T/G > A calls were removed in two FFPE Twist samples, P2 and P3, and in one FFPE Agilent sample, P1. In C > A/G > T mutations, the fraction of the artifact varied greatly across the samples, from 5.5 to 71.2%. The C > A/G > T artifact is mainly ascribed to the formation of 8-oxoguanine, a type of DNA damage caused by oxidative stress that mostly occurs during DNA shearing process19. Consistently, the Twist FFPE sample, P3, that showed the highest proportion of the C > A/G > T artifact (71.2%) exhibited notably shorter insert sizes (Fig. 2c and d), suggesting that an additional DNA oxidative damage might have occurred in the sample.
Next, we assessed the concordance of variant allele frequency of SNV calls (depth ≥ 20) that were commonly detected in current and previous studies (Fig. 3a). All Pearson’s correlation coefficients were ≥ 0.91, ranging from 0.914 to 0.938, indicating that the commonly detected calls are reliable. We further examined if the somatic SNV calls from the previous study were successfully reproduced in the current study. Since many artifacts were detected in primary calls (Fig. 2e and f), we employed four sequential filtering methods: GATK Mutect2 (FilterMutectCalls), SOBDetector9, FFPolish8, and MicroSEC7. Assuming that the SNV calls detected in both studies are true variants, FFPolish appeared to eliminate artifacts most effectively while preserving most of the true variants, suggesting that filtration with FFPolish profoundly increased sensitivity with little loss of specificity (Fig. 3b and c). After four filtering steps, 83.0–97.0% of filtered somatic SNV calls from the previous study were recovered in the current study in four FFPE samples at the level of alternate AF > 0 (Table S4 and S5). As the AF increased, the proportion of the SNV calls that were not validated in the reciprocal study decreased. This is congruent with a previous finding that many low-frequency SNVs detected in FFPE samples were false positives9. On the other hand, much higher numbers of false positive calls were observed in the current study, which might be attributed to the accelerated introduction of additional DNA damages in the vulnerable DNA samples which were already compromised by pre-existing damages due to prolonged storage. Furthermore, single strand-based libraries are more likely to capture the damaged DNA molecules17,18, resulting in increase of artifactual calls in current study. In addition, single-stranded DNA molecules in FFPE can produce artifact chimeric reads and lead to subsequent sequence errors22,23.

Evaluation of exome seqeuncing protocol. (a) Scatter plots of variant allele frequencies of SNV calls (depth ≥ 20) from current study (Twist) and those from previous study (Agilent) in nine pediatric brain caner samples with regression lines and Pearson’s correlation coefficients. (b) Number of validated and not-validated Agilent (previous study) SNV calls after four sequential filtering steps. (c) Number of validated and not-validated Twist (current study) SNV calls after four sequential filtering steps. (d) Comparison of genotype calls of NA12878 (depth ≥ 10) between current study (Twist) and public datasets from the Genome-in-a-Bottle (GIAB) consortium (HG001) and GSE206253. (e) Comparison of genotype calls of NA12878 (depth ≥ 10) after filtration of the calls of current study (Twist) using GATK CNNScoreVariants 1D and 2D model.
Subsequently, we compared genotype calls from the reference NA12878 sample to those of two public datasets from the Genome-in-a-Bottle (GIAB) consortium (HG001)12 and GSE206253. Total 19,029 variant calls were commonly detected in three datasets with a minimum depth of 10x (Fig. 3d). After filtration of the genotype calls of current study (Twist) by GATK CNNScoreVariants 1D and 2D models with default options, total 18,373 calls were commonly recognized in the three datasets (Fig. 3e).
Evaluation of performance of methylome sequencing
Bisulfite conversion rate estimated from percentages of cytosine methylation in CHG and CHH (H = A, C, or T) contexts was ≥ 99% in all samples (Table S6). Basic statistics of methylome sequencing data are summarized in Table S7. In nine pediatric brain cancer samples, the proportion of uniquely aligned reads ranged 77.2–83.5%, with duplication rates of 11.3–41.1%. The average off-target rate across all samples was 14.1%, ranging from 7.0 to 33.1%. The average of fold 80 base penalty, a metric for evenness of coverage, was 2.02, ranging from 1.65 to 2.73. Median insert size was relatively small in three FFPE samples (P1 - P3) (Fig. 4a), indicating that additional DNA degradation occurred in the three samples.

Evaluation of methylome sequencing protocol. (a) Median insert size calculated from uniquely aligned properly paired reads (mapping quality ≥ 20). (b) Number of detected CpG sites according to depth of coverage. (c) Number of CpG sites detected at 1x, 5x, and 10x. (d) Pairwise Pearson’s correlation coefficients cacluated with methylation values on strand-specific 60,187 autosomal CpG sites commonly detected in current and three public datasets (depth ≥ 10). (e) Pairwise Pearson’s correlation coefficients calculated with 300,904 autosomal CpG sites commonly detected in nine pediatric brain cancer samples (depth ≥ 10) after excluding the CpG sites where primary somatic SNV calls were observed. (f) Inverse correlation between Pearson’s correlation coefficients calculated with methylome profiles and average number of artifactual variants in three pairs of matched samples. (g) Distibution of methylation levels in four reference HCT116 gDNA samples with 100%, 70%, 30%, and 0% methylation proportion (depth ≥ 10).
On average, approximately 3.22 million CpGs were identified with at least 10x depth, ranging from 1.01 to 3.90 million CpGs across all samples (Fig. 4b, 4c, and Table S8). The number of detected CpGs in three FFPE samples P1, P3, and P4 were comparable to those detected in reference gDNA samples or fresh frozen samples. The least detection number of CpGs, approximately 1.01 million, was observed in FFPE sample P2, for which a lower input amount of DNA (33.4 ng) was applied for construction of methylome library (Table 1). Most of the methylome sequencing statistics of the P2 sample were significantly inferior including total raw read count and median depth of coverage (Table S7). In fact, the sample P2 appeared to be most severely damaged among the FFPE samples, as evident from the gel electrophoresis (Figure S1) and the estimated median insert size (Fig. 4a). This implies that the initial severe damage in P2 might have been exacerbated by bisulfite treatment, resulting in further DNA degradation and length reduction, which eventually led to hybridization failure with the probes of Twist methylome panel with probe length of 120 bp and finally caused diminished total read count and detection number of CpGs.
To further evaluate the accuracy of estimated DNA methylation levels, the extracted methylation values of NA12878 were compared with those from three publicly available whole-genome bisulfite sequencing (WGBS) datasets. Pearson’s correlation coefficients were calculated considering strand-specific 60,187 autosomal CpG sites that were commonly detected in all four datasets with depth of at least 10x within the target region of Twist methylome panel. The observed pairwise correlation coefficients ranged from 0.933 to 0.935, which was close to the correlation level, 0.944, calculated between two replicate samples, GSM6348305 and GSM6348306 (Fig. 4d). We further calculated pairwise correlation coefficients based on 300,904 autosomal CpG sites that were commonly detected in nine pediatric brain cancer samples with a depth of at least 10x. The results showed high correlations, ranging from 0.90 to 0.97, in three pairs of matched samples from the same donors (Fig. 4e). Furthermore, we observed an inverse correlation between the Pearson’s correlation coefficients calculated from methylation profiles and the average number of artifactual SNV calls detected in the result of Fig. 2e (Fig. 4f), suggesting that the number of artifactual calls estimated from exome data could serve to infer the inaccuracy of methylation estimation. However, a small sample size may not provide a definitive conclusion and further studies with larger sample sizes are needed to confirm the relationship. Finally, density plots of methylation values in four HCT116 reference samples illustrated good concordance between the expected methylation levels and estimated ones, indicating high performance of our methylome protocol (Fig. 4g).
SNV calling from methylome data
Based on a recently developed bioinformatics tool for simultaneous extraction of genetic and epigenetic information from DNA methylation sequencing data11, we generated SNV calls from methylome data and compared them with those from exome data in the common target region (~ 10.6 Mb) of Twist Exome 2.0 Panel and Twist Human Methylome Panel. In nine clinical samples, 39–76% of filtered SNV calls from exome data were recovered by methylome data (Fig. 5a) and the correlation coefficient of variant allele frequency of the commonly detected SNVs ranged from 0.175 to 0.692 (Fig. 5b). When considering huge number of false positive calls detected from methylome data, mutation analysis based on methylation sequencing data may not be practical. However, it can complement exome sequencing data in variant detection.

SNV calls from exome data recovered by methylome data in the common target region of Twist Exome 2.0 Panel and Twist Human Methylome Panel. (a) Comparison of SNV calls from exome and methylome data. Numbers in white color indicate SNVs identified from exome data after four filtering steps. (b) Correlation between variant allele frequencies of SNV calls (depth ≥ 10) from exome data and those from methylome data in nine pediatric brain caner samples with regression lines and Pearson’s correlation coefficients.
Discussion
Numerous studies have shown that integrated analysis of combined omics data provides multi-dimensional information from the original cause of disease to the functional consequences or relevant interactions23–25. Combined use of genetic and epigenetic markers also provides advantages in clinical fields such as an enhanced diagnostic power for prediction of prognosis3. Therefore, simultaneous acquisition of genomic and epigenomic information from a single DNA sample in a rapid and simple way will provide great benefits in both clinical and research settings. In this regard, we established a fast and efficient protocol for parallel construction of exome and methylome libraries, which enabled us to obtain targeted exome and methylome NGS libraries within 1.5 days with a quite small amount of initial input DNA. Furthermore, reliable targeted genomic and epigenomic data were produced even from the DNA samples containing a wide range of damages from FFPE processing, oxidation of DNA, and/or long-term storage. In general, the construction of exome and methylome sequencing libraries typically employs distinct protocols, reagents, and techniques. In contrast, we applied a unified workflow using the same reagents and techniques for both exome and methylome, which ultimately reduced complexity and variability as well as library preparation time by simplifying the process.
In methylation sequencing, the overall distribution of sequenced bases can be uneven due to bisulfite-converted bases. Therefore, Illumina, for example, recommends incorporating PhiX in most library preparations as a quality and calibration control for sequencing runs. In this study, the exome library with a balanced base composition and the methylome library with an unbalanced base composition were sequenced simultaneously within a single lane on the Illumina platform. As a result, the exome library provided a correction effect while maintaining an overall uniform base rate. Therefore, there was no need to add additional PhiX for signal correction, resulting in a 5–20% reduction in sequencing costs. In addition, genetic alterations can be detected from methylome data, which can provide simultaneous validation of genetic variation and/or increase overall depth of coverage for mutation calls, and ultimately reduces total sequencing costs.
In recent studies, several new technologies have been developed to obtain genetic and epigenetic information simultaneously from a single DNA molecule14,16,26. These new strategies utilize complementary information of both strands of DNA by ligation of hairpin adapters to the ends of double-stranded DNAs. In one of the studies, the new methodology was applied to small amounts of circulating free DNA (cfDNA)16. However, the technology has not been tested for a target enrichment approach yet. In another study, the new technology was successfully employed for target enrichment, however, it was achieved by using a high amount of DNA14. Although our protocol did not delineate such a novel sequencing technology extracting genetic and epigenetic information simultaneously from a single DNA molecule, we demonstrated that it can be successfully applied to a small amount of damaged DNA with a target enrichment approach. In addition, we used commercially available materials in all processes, which allows for easy access and application in both research and clinical fields.
In summary, we present a comprehensive and efficient workflow utilizing a single-strand based DNA library preparation method to obtain NGS libraries specifically designed for exome and methylome analysis, which can be applied to even a small amount of damaged DNA sample. Based on the protocol from this study, narrowing down the scope of targets could lead to the development of clinical panels for simultaneous detection of genetic and epigenetic alterations, which is applicable to a wide range of DNA samples including those from rare and old clinical samples. However, sequencing artifacts can arise from various sources such as significant damage to nucleic acids or chimeric reads derived from single-stranded DNA molecules7,22. Single strand-based libraries are more prone to incorporating damaged and chimeric DNA molecules, leading to an increased rate of misidentification of genetic variants, which must be detected and removed by employing appropriate algorithms and filtering steps. In addition, although our protocol may be useful for exon regions that have many unique sequences, it may not be appropriate for sequencing a whole genome region that has many repeats and homologous sequences due to its small insert size.
Data availability
The exome and methylome sequence data generated in current study have deposited into sequence read archive (SRA) database under accession number PRJNA1144056. Publicly available exome datasets of pediatric medulloblastoma were downloaded from PRJNA418669 in the SRA database. Two public VCF files of NA12878, HG001 and GSE206253, were downloaded from https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/NISTv4.2.1/GRCh38/HG001_GRCh38_1_22_v4.2.1_benchmark.vcf.gz and the gene expression omnibus (GEO) database, respectively. Three public CpG methylation call datasets of NA12878, GSM2772524, GSM6348305, and GSM6348306, were downloaded from the GEO database.
References
Chakraborty, S., Hosen, M. I., Ahmed, M. & Shekhar, H. U. Onco-multi-OMICS approach: A new frontier in cancer research. Biomed. Res. Int. 2018, 9836256. https://doi.org/10.1155/2018/9836256 (2018).
Levy, S. E. & Myers, R. M. Advancements in Next-Generation sequencing. Annu. Rev. Genomi. Hum. Genet. 17, 95–115. https://doi.org/10.1146/annurev-genom-083115-022413 (2016).
Parikh, A. R. et al. Minimal residual disease detection using a Plasma-only Circulating tumor DNA assay in patients with colorectal Cancer. Clin. Cancer Res. 27, 5586–5594. https://doi.org/10.1158/1078-0432.CCR-21-0410 (2021).
Arreaza, G. et al. Pre-Analytical considerations for successful Next-Generation sequencing (NGS): challenges and opportunities for Formalin-Fixed and Paraffin-Embedded tumor tissue (FFPE) samples. Int. J. Mol. Sci. 17. https://doi.org/10.3390/ijms17091579 (2016).
Ludgate, J. L. et al. A streamlined method for analysing genome-wide DNA methylation patterns from low amounts of FFPE DNA. BMC Med. Genomi. 10, 54. https://doi.org/10.1186/s12920-017-0290-1 (2017).
Phi, J. H. et al. Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent Medulloblastomas. Acta Neuropathol. 135, 939–953. https://doi.org/10.1007/s00401-018-1845-8 (2018).
Ikegami, M. et al. MicroSEC filters sequence errors for formalin-fixed and paraffin-embedded samples. Commun. Biol. 4, 1396. https://doi.org/10.1038/s42003-021-02930-4 (2021).
Dodani, D. D., Nguyen, M. H., Morin, R. D., Marra, M. A. & Corbett, R. D. Combinatorial and machine learning approaches for improved somatic variant calling from Formalin-Fixed Paraffin-Embedded genome sequence data. Front. Genet. 13, 834764. https://doi.org/10.3389/fgene.2022.834764 (2022).
Diossy, M. et al. Strand orientation Bias detector to determine the probability of FFPE sequencing artifacts. Brief. Bioinform. 22. https://doi.org/10.1093/bib/bbab186 (2021).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. https://doi.org/10.1093/bioinformatics/btr167 (2011).
Zhou, W. et al. BISCUIT: an efficient, standards-compliant tool suite for simultaneous genetic and epigenetic inference in bulk and single-cell studies. Nucleic Acids Res. 52, e32. https://doi.org/10.1093/nar/gkae097 (2024).
Płoski, R. Next Generation Sequencing-General Information about the Technology, Possibilities, and Limitations in Clinical Applications for Next-Generation Sequencng, (ed. Demkow, U., Płoski, R), 1–18 (Academic, 2016).
Zook, J. M. et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 37, 561–566. https://doi.org/10.1038/s41587-019-0074-6 (2019).
Yan, B., Wang, D., Vaisvila, R., Sun, Z. & Ettwiller, L. Methyl-SNP-seq reveals dual readouts of methylome and variome at molecule resolution while enabling target enrichment. Genome Res. 32, 2079–2091. https://doi.org/10.1101/gr.277080.122 (2022).
Suzuki, M. et al. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome Res. 28, 1364–1371. https://doi.org/10.1101/gr.232587.117 (2018).
Fullgrabe, J. et al. Simultaneous sequencing of genetic and epigenetic bases in DNA. Nat. Biotechnol. 41, 1457–1464. https://doi.org/10.1038/s41587-022-01652-0 (2023).
Burnham, P. et al. Single-stranded DNA library Preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 6, 27859. https://doi.org/10.1038/srep27859 (2016).
Vong, J. S. L. et al. Single-Stranded DNA library Preparation preferentially enriches short maternal DNA in maternal plasma. Clin. Chem. 63, 1031–1037. https://doi.org/10.1373/clinchem.2016.268656 (2017).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample Preparation. Nucleic Acids Res. 41, e67. https://doi.org/10.1093/nar/gks1443 (2013).
Do, H. & Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 61, 64–71. https://doi.org/10.1373/clinchem.2014.223040 (2015).
Oh, E. et al. Comparison of accuracy of Whole-Exome sequencing with Formalin-Fixed Paraffin-Embedded and fresh frozen tissue samples. PLoS One 10, e0144162. https://doi.org/10.1371/journal.pone.0144162 (2015).
Haile, S. et al. Sources of erroneous sequences and artifact chimeric reads in next generation sequencing of genomic DNA from formalin-fixed paraffin-embedded samples. Nucleic Acids Res. 47, e12. https://doi.org/10.1093/nar/gky1142 (2019).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83. https://doi.org/10.1186/s13059-017-1215-1 (2017).
Subramanian, I., Verma, S., Kumar, S., Jere, A. & Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform Biol. Insights 14, 1177932219899051. https://doi.org/10.1177/1177932219899051 (2020).
Chai, P., Yu, J., Ge, S., Jia, R. & Fan, X. Genetic alteration, RNA expression, and DNA methylation profiling of coronavirus disease 2019 (COVID-19) receptor ACE2 in malignancies: a pan-cancer analysis. J. Hematol. Oncol. 13, 43. https://doi.org/10.1186/s13045-020-00883-5 (2020).
Liang, J. et al. A new approach to Decode DNA methylome and genomic variants simultaneously from double strand bisulfite sequencing. Brief. Bioinform. 22. https://doi.org/10.1093/bib/bbab201 (2021).
Acknowledgements
We thank Ji Yoon Lee’s families for their donation to pediatric brain tumor research at the Seoul National University Children’s Hospital (3020190190). We also wish to acknowledge all researchers who provided the data.
Funding
This work was supported by Industry-university Cooperation Collabo R&D Program funded by the Ministry of SMEs and Startups, Republic of Korea (RS-2023-00226999), a National Research Foundation (NRF) grant of Korea funded by the Ministry of Science & ICT (2020R1I1A3073845), SNUH Kun-hee Lee Child Cancer & Rare Disease Project, Republic of Korea (grant number: 22 A-017-0100), and research funds for newly appointed professors of Jeonbuk National University in 2022.
Author information
Authors and Affiliations
Contributions
A.K.P., S.A., and S-K.K. conceived and supervised the research. S.A. and H.A. designed and performed the wet lab experiments. A.K.P. and E.K. analyzed the data. J.L. supported the analysis and interpretation of data. A.K.P., S.A., and E.K. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the institutional review board of Jeonbuk National University (IRB JBNU-2023-05-025).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, E., An, S., Ahn, H. et al. Fast and efficient method for parallel construction of targeted exome and methylome single-stranded DNA sequencing libraries. Sci Rep 15, 7144 (2025). https://doi.org/10.1038/s41598-025-91537-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-91537-4
