
Abstract
As an infectious disease that poses a significant threat to the rapidly growing pig breeding industry, the detection of Haemophilus parasuis (HPS) is often compromised by various interfering substances present in the test sample during quantitative real-time PCR (qPCR). The rapid detection of HPS is important for the isolation of infectious pigs and their treatment. We designed and optimized a rapid qPCR test to detect the INFB gene of HPS in clinical and environmental samples on pig farms. The method was evaluated for its specificity, sensitivity, repeatability, anti-interference capability, and its ability to detect HPS in clinical samples. The results indicated that the method was specific for the detection of HPS when evaluated against pathogens and intestinal probiotics found in pig farms. By using a seven-fold dilution series of the recombinant plasmid DNA in triplicate, it was determined that the lowest limit of detection (LOD) for this method was less than 10 copies/µL. The results of inter-batch and intra-batch repeatability tests showed that the coefficient of variation (CV) was consistently below 1%. Furthermore, the impact of 14 endogenous and exogenous interfering substances on the Ct values detected by the HPS qPCR was found to be less than 5% when compared to the Ct values obtained in the absence of interfering substances. A total of 248 clinical samples were analyzed using the HPS qPCR, commercial kits, and corresponding national standards, yielding positive rates of 9.27%, 6.05%, and 9.27%, respectively. Notably, the positive and negative percent agreement between the detection method developed in this research and the national standard was 100%. These findings demonstrate that the established detection method is suitable for epidemiological research on HPS and for diagnosing clinical samples containing interfering substances, thereby providing essential technical support for the prevention and control of HPS.
Similar content being viewed by others

Introduction
Haemophilus parasuis disease (HPSD), also known as polyfibrinous serositis and arthritis or Glaesser’s disease, is an infection in animals caused by Haemophilus parasuis(HPS) with pigs as the exclusive host1,2. In recent years, H. parasuis has been reclassified as Glaesserella parasuis3. Clinical signs of HPSD include fever, joint inflammation, breathing difficulties,and the presence of multiple serositis and arthritis4,5. This disease primarily affects pigs before and after weaning, as well as during the nursery stage, with an incidence rate ranging from 10–15%6. In cases of severe acute HPSD, the mortality rate in pigs can reach up to 50%, posing a significant threat to the health of piglets and young pigs7. The likelihood of the disease occurring increases when there are other infections such as reproductive respiratory syndrome, influenza, or enzootic pneumonia within the herd8,9,10. Due to the ubiquitous nature of HPS in the environment and its presence in both healthy and diseased pigs, HPS is a global issue that seriously impacts the swine industry11,12,13.
HPS is a gram-negative bacterium with variable morphology and over 15 serotypes, with serotypes 5, 4, and 13 being the most prevalent, comprising more than 70% of cases14,15,16. Approximately 20% of clinical strains remain untyped17. Variability in virulence and pathogenicity exists among different serotypes of HPS, with even strains of the same serotype demonstrating varying levels of pathogenicity and incidence rates5,18. The virulence of strains is closely linked to the virulence genes they harbor. Serotypes 1, 5, 10, 13, and 14 are highly virulent, while types 2, 4, and 15 are moderately virulent; other serotypes are considered non-virulent5,18,19. Extensive research has been conducted on the pathogenic mechanisms of HPS, immune response induction, and the development of vaccines. However, due to frequent co-infections with other pathogens and weak cross-protection between serotypes, vaccine efficacy is limited, resulting in widespread disease outbreaks that significantly impact the pig industry2,20. The rapid identification and physical isolation of sick pigs are the most effective methods for preventing and controlling the spread of HPSD. Consequently, there is an urgent need to develop rapid detection technologies for HPS.
Various methods have been developed for detecting HPS, including serological and molecular biology techniques21,22,23. Serological methods, including the complement fixation test, indirect hemagglutination test and enzyme-linked immunosorbent assay, exhibit limitations such as low analytical sensitivity and weak specificity during the early stages of pathogen infection24,25,26,27. In contrast, molecular biology methods like polymerase chain reaction (PCR), real-time fluorescence quantitative PCR (qPCR), loop-mediated isothermal amplification (LAMP), and nucleic acid molecular hybridization offer higher specificity and sensitivity. Among them, qPCR is the most widely used in virus detection. However, current qPCR methods for HPS detection lack specificity as they are based on the 16 S rDNA sequence common to bacteria of the genus Haemophilus, leading to potential false positives28. Currently, existing HPS qPCR detection methods exhibit low sensitivity and may not be suitable for identifying low bacterial loads. Furthermore, the complexity of sample components significantly impacts the stability of the HPS qPCR reaction system. During the sample collection process, various exogenous and endogenous components are often introduced. These interfering substances, whether derived from the individual or the external environment, can interact with nucleic acids and impede the binding of primers to their templates. Additionally, these substances may directly inhibit the activity of enzymes involved in PCR reactions, thereby obstructing the normal progression of the reaction29,30.
Based on the above considerations, this study developed primers and probes based on the conserved region of the HPS INFB gene. By screening primers and optimizing the reaction system, a highly specific, sensitive, and low coefficient of variation HPS qPCR detection method was established. The HPS qPCR system described in this study is capable of rapidly detecting HPS in complex samples with low bacterial load, offering a new technical approach for the prevention and diagnosis.
Materials and methods
Samples and materials
The nucleic acids employed to assess the specificity of our method for detecting HPS in clinical samples were extracted from a range of viral vaccines and bacteria in this study. These included the porcine mycoplasma pneumonia inactivated vaccine, porcine atrophic rhinitis inactivated vaccine (Bordetella JB5 strain), porcine parvovirus inactivated vaccine (WH-1 strain), Porcine circovirus disease type 2 inactivated vaccine (SH strain), swine fever, swine erysipelas, porcine pasteurellosis multocida triple live vaccine, porcine pseudorabies live vaccine (HB-98 strain), Enterococcus faecalis, and Clostridium butyricum which were purchased from vaccine companies and probiotic companies. Detail information can be found in Supplement Table S1. Additionally, Escherichia coli, Bacillus subtilis, and Salmonella were isolated and stored in our laboratory. The analytical grade ethanol and isopropanol (purity ≥ 99.7%) utilized in this experiment were acquired from Samson Chemical Technology (Shanghai) Limited Company.
Samples were collected from pigs exhibiting clinical symptoms of HPS, as well as from their living environments in pig raising enterprises located in Jilin Province. These samples were obtained through random sampling. The total number of clinical samples analyzed was 248, comprising 56 whole blood samples, 48 serum samples, 32 swab samples from the oral cavity, nose, and anus, and 74 tissue samples (lung, spleen, liver, kidney, tonsil, and lymph node). Additionally, there were 26 saliva samples and 12 environmental samples, all of which were stored at 4℃ for future use.
Nucleic acid extraction
Nucleic acids were extracted from vaccines, constructed plasmids, and clinical samples using the magnetic bead bacterial total nucleic acid extraction kit and the magnetic bead viral total nucleic acid extraction kit, both purchased from Sairuisi Biotechnology Co., Ltd. (Jilin, China). For blood and faeces, the volume of the lysate or duration of water bath incubation was amended according to kit instructions for nucleic acid extraction for these sample types. The extracted nucleic acids were then stored at −20 °C for future use.
Primer and probe design
Primer and probe design utilized the published GenBank numbers of 15 HPS serotypes randomly obtained from NCBI. The registration numbers and corresponding serotypes of HPS are shown in Table 1. Utilize Vector NTI Advance (11.5.1) to align the downloaded gene sequences and design primers (Figure S1). The designed primers and probes were then evaluated using PrimeSelect software to assess physical parameters, including annealing temperature, hairpin structure, primer-dimer formation, and free energy. The specificity of the primers and probes was verified using the BLAST tool provided by NCBI (http://www.ncbi.nlm.nih.gov/blast). Subsequently, primers with similar annealing temperatures, high specificity, low free energy, and minimal formation of primer dimers and hairpin structures were selected for synthesis. The cytoskeletal housekeeping gene, β-actin, is expressed in pig cells and was used as an internal control to ensure nucleic acids were recovered from porcine samples. The synthesis of primers and probes was conducted by Sangon (Shanghai, China). The sequences of the primers and probes are detailed in Table 2.
Procedures and systems of amplification reaction
In this study, three PCR methods were employed: conventional PCR, SYBR Green I qPCR, and TaqMan qPCR. The primary function of conventional PCR is to amplify the target fragment, which is subsequently purified using gel electrophoresis and utilized for plasmid construction. The primers utilized in this process are HPS-F1/R1, as listed in Table 1. Both SYBR Green I qPCR and TaqMan qPCR were implemented to evaluate the amplification efficacy of the two primer pairs, HPS-F1/R1 and HPS-F2/R2, also detailed in Table 2. The amplification reaction systems, amplification conditions, and the concentrations of primers and probes used for the three PCR methods are outlined below.
The conventional PCR system used in this study involved combining 2 x PCR Master Mix (Sangon, Shanghai, China) 10 µL, 0.5 µL each of forward and reverse primers (10 µM), 2 µL template, and ddH2O to make up to a total volume of 20 µL. The PCR amplification program included an initial pre-denaturation step at 95℃ for 5 min, followed by 40 cycles of denaturation at 95℃ for 30 s, annealing at 55℃ for 30 s, extension at 72℃ for 45 s, and a final extension at 72℃ for 10 min. This programme is used for plasmid construction.
The SYBR Green I qPCR reaction system consisted of 2 x RealStar Fast dye qPCR premix (UNG, High ROX) (Kangrun, Beijing, China) 10 µL, 0.5 µL each of the forward and reverse primers (10 µM), DNA template 1 µL, and ddH2O to a final volume of 20 µL. The reaction protocol included UNG enzyme treatment at 50℃ for 5 min, followed by pre-denaturation at 95℃ for 2 min, and 40 cycles of reaction (denaturation at 95℃ for 15 s, annealing at 60℃ for 30 s). The melting curve analysis was performed automatically using a StepOnePlus real-time fluorescence quantitative PCR instrument (Thermo Fisher, Waltham, MA, USA). Thresholds are set automatically by the machine.
TaqMan qPCR was performed using 2xTaq Pro U+ Multiple qPCR Mix (Nazyme, Nanjing, China), with 10 µL total volume. The reaction mixture included 0.2 µL each of forward and reverse primers (10 µM), 0.1 µL probe (10 µM), 0.4 µL of 50x ROX Reference Dye, 2 µL of DNA template, and ddH2O to a final volume of 20 µL. The reaction program consisted of a contamination digestion step at 37℃ for 2 min, followed by pre-denaturation at 95℃ for 30 s, and 40 cycles of reaction (denaturation at 95℃ for 10 s, annealing at 60℃ for 30 s). The amplification products generated during repeated qPCR amplification are a primary source of aerosol contamination of nucleic acid amplicons. This aerosol contamination is a significant contributor to false positive detection results. To mitigate the risk of false positive HPS results caused by these nucleic acid amplicon aerosols, a UNG enzyme anti-contamination system was incorporated into the TaqMan qPCR amplification system.
Recombinant plasmid Preparation
The genomic DNA of the HPS strain was amplified using a selected primer set (HPS-F1/R1) through PCR. Following PCR amplification, the product underwent 2% agarose gel electrophoresis to isolate the target band, which was then ligated with the pMD19-T plasmid vector (Takara, Beijing, China) and introduced into E. coli DH5α competent cells (Sangon, Shanghai, China). Enrichment culture and screening of positive clones were carried out. The success of plasmid construction was determined by PCR amplification and sequencing results. The successfully constructed positive plasmid was named pMD19-INFB and stored for future use. Nucleic acids from the positive plasmid pMD19-INFB were extracted according to the method described in Sect. 2.2, and subsequently, the gene copy number corresponding to 1 ng of DNA was calculated using the following formula.
copies/mL = 6.02 × 1023(copies/mol) × (concentration g/mL) / (MW g/mol)32,33.
Primer and probe selection
Using the DNA from the recombinant plasmid pMD19-INFB as the amplification template, both the SYBR Green I staining method and the TaqMan probe method were employed to screen the primers and probes listed in Table 2. The absence of primer-dimers in the melting curve and low Ct values were utilized as criteria for screening HPS primer probes.
Optimization of HPS qPCR system
In order to determine the optimal qPCR reaction conditions, the primer, probe concentrations, and annealing temperature were optimized using plasmid DNA templates at concentrations of 3.16 × 103 copies/µL, 3.16 × 104 copies/µL, and 3.16 × 105 copies/µL. The primer volume range tested were 50 nM, 100 nM, and 150 nM (with the probe concentration held constant at 50 nM). After identifying the optimal primer concentration, probe volume optimization was conducted with levels of 50 nM, 100 nM, and 150 nM. Subsequently, annealing temperature optimization was carried out with temperatures of 58 °C, 60 °C, and 62 °C. Each experiment was repeated three times.
Specificity test
In this study, 11 pathogens commonly found in swine farms were selected for specificity testing. These pathogens included Streptococcus suis (SS), Escherichia coli (ED), Erysipelas reinhardtii (Ery), Actinobacillus pleuropneumoniae (APP), Mycoplasma pneumoniae (Mp), Salmonella porcine, Pasteurella porcine (PRDC), infectious rhinitis of pig (AR), pseudorabies virus (PRV), porcine papillomavirus (PPV), and porcine circovirus 2 (PCV2). Additionally, nucleic acids of three porcine companion probiotics (Clostridium butyricum, Enterococcus faecalis, and Bacillus subtilis) were also used for specificity validation of the HPS TaqMan qPCR method. The specificity experiments involved using a standard plasmid with 3.16 × 106 copies/µL as a positive control and ddH2O as a negative control. The amplification process followed optimized reaction conditions and was repeated three times for accuracy.
Establishment of standard curves
The recombinant plasmid pMD19-INFB was utilized as a qPCR template after being diluted 10-fold to a concentration ranging from 102 copies/µL to 106 copies/µL. The optimized reaction system and conditions were employed for a single qPCR amplification, and a standard curve was generated by plotting the logarithm of the initial copy number concentration of the plasmid standard against the Ct value.
Lowest limit of detection (LOD)
To determine the lowest detection limit of the established HPS qPCR, we diluted the plasmid DNA to a concentration range of 3.16 × 107 copies/µL to 3.16 × 100, and utilized an optimized final qPCR reaction system for amplification. The minimum plasmid concentration that successfully generated an amplification curve was defined as the minimum detection limit. Each concentration was tested in triplicate to ensure the accuracy of the experiment. Additionally, ddH2O was utilized as a no template control during the amplification process to validate the reliability of the experimental procedure. Subsequently, the sensitivity results of the qPCR assay guided the selection of an appropriate concentration range for conventional PCR amplification. In addition, the performance of the HPS qPCR reaction system was further assessed by comparing it with commercially available kits(Weiboxin, Guangzhou, China) using genomic DNA extracted from HPS vaccines, which were subjected to a tenfold gradient dilution as a template.
Precision test
To assess the repeatability of the HPS qPCR assay system, qPCR amplification was conducted with standard plasmids at three different concentrations (3.16 × 103 copies/µL, 3.16 × 104 copies/µL, and 3.16 × 105 copies/µL) to evaluate the intra- and inter-batch precision of the experiments. Intra-batch precision was assessed by using the same batch of prepared qPCR reaction systems and performing three replicate amplifications for each template concentration. Inter-batch precision was evaluated by preparing 5 batches of qPCR reaction systems over 5 working days and conducting 3 amplifications for each template concentration. The coefficient of variation (CV) was calculated as the standard deviation (SD) divided by the mean, multiplied by 100%. If the coefficient of variation (CV) value is lower than 5%, the established HPS qPCR detection system is deemed to exhibit good repeatability.
Interference test
The HPS assay samples are often collected in complex environments, where they may contain various interfering substances that can greatly impact the sensitivity and reliability of the assay results. To assess the tolerance of the HPS qPCR assay system to potential interfering substances, an experimental protocol was developed based on the specific clinical samples and potential interferers present. In this protocol, 100 µL of a medium positive plasmid standard (3.16 × 105 copies/µL) was mixed with either 8 endogenous interfering substances or 6 exogenous interfering substances as the amplification template, and an optimized qPCR system was utilized for amplification. Endogenous interfering substances refer to physiological components present in the test sample that can interfere with substance analysis, including mucins, latex, blood, heart, liver, spleen, lungs, and intestine. In contrast, exogenous interfering substances are those originating from outside the body that can also affect substance analysis, including Reagent 1–4, feces and feeds. Additionally, a positive control consisting of 100 µL of medium-positive plasmid standard mixed with 100 µL of ddH2O was included, along with a negative control of 200 µL of ddH2O to assess the impact of potential interferers on the assay. Each set of experiments was conducted in triplicate, and the final results were reported as the mean Ct value ± standard deviation (SD). An increase of 5% in the Ct value of the interference group compared to the positive control is classified as slight inhibition, whereas an increase of 10% is deemed significant inhibition.
Testing of clinical samples
DNA was extracted from the clinical samples described in Sect. 2.1, and HPS was detected using the method established in this study, along with the purchased HPS nucleic acid detection kit (Wei Boxin, Guangzhou, China). The national standard for HPS detection, as outlined in SN/T4230-2015 “Technical Specifications for Quarantine of Haemophilus parasuis,” served as the gold standard for evaluating the detection efficacy of the two methods. To ensure the accuracy of the test results, the positive control provided in the purchased kit was utilized to confirm successful amplification. Additionally, the entire extraction process was monitored using the porcine housekeeping gene β-actin. The amplification process was controlled with water as a no template control, and an open tube control was employed to evaluate the extraction environment. This comparison was conducted using three indicators: positive percent agreement, negative percent agreement and Cohen’s Kappa. The calculation method for positive and negative percent agreement involves determining the number of samples classified as positive or negative by both the HPS qPCR (commercial kit) and the national standards. This value is then divided by the number of samples classified as positive or negative by the national standards alone, and the result is multiplied by 100%.
Results
Results of primer probe screening
To improve amplification, primers were initially screened using both SYBR Green I and TaqMan probe methods. Melting curves indicated that HPS-F1/R1 and HPS-F2/R2 were devoid of Primer dimers and non-specific amplification (Fig. 1A). Further screening based on Ct value revealed that HPS-F1/R1 showed superior amplification results with both SYBR Green I and TaqMan probe methods (Fig. 1B). Consequently, HPS-F1/R1 and its corresponding probe were chosen for subsequent experiments.

(A) Melting curves of HPS-F1/R1 and HPS-F2/R2; (B) Comparison of amplification effects of HPS-F1/R1 and HPS-F2/R2.
Identification of plasmid pMD19-INFB
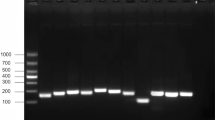
Figure 2A illustrates the constructed recombinant plasmid. The PCR amplification product of the plasmid DNA ranged from 100 to 250 bp, confirming the successful construction of the plasmid (Fig. 2B). Additionally, sequencing results further validated the successful construction of the plasmid. Consequently, this constructed plasmid is suitable for use in subsequent experiments. Furthermore, based on the copy number calculation formula, 1 ng of plasmid DNA corresponds to approximately 3.16 × 109 copies/µL.

(A) pMD19-INFB plasmid construction; (B) Gel electrophoresis results of PCR amplification products; M is Marker with molecular weight 2000, 1 and 2 are amplification products.
Optimization of HPS qPCR reaction system
To identify the most effective reaction system for HPS qPCR detection, this study systematically optimized key conditions using three different concentrations of recombinant plasmid DNA. The results are detailed in Table 3. It was found that with a primer concentration of 150 nM, probe concentration of 100 nM, and an annealing temperature of 60 °C, the reaction system demonstrated the most efficient amplification. This optimized qPCR reaction system can now be utilized for future experiment.
Specificity of HPS qPCR
In order to validate the specificity of the HPS qPCR method, nucleic acids were extracted from 11 common pathogens found in pig farms and 3 types of pig-associated probiotics. Subsequently, qPCR amplification was performed. As shown in Fig. 3, the detection method established in this study demonstrated specific amplification solely for HPS nucleic acid samples, without showing specific amplification for 11 common pathogens and 3 pig-related probiotics. Consequently, the qPCR detection method designed in this study exhibited strong specificity in detecting common pig pathogens.

HPS qPCR specificity assay.
Establishment of HPS qPCR standard curve
The standard curve of HPS positive plasmid is illustrated in Fig. 4, showing a linear relationship between plasmid copy number and Ct value with the equation y = −3.415x + 19.669. The correlation coefficient (R2) is 0.997, and the amplification efficiency is 96.26%. These results indicate that the qPCR assay for HPS developed in this study exhibits a strong linear relationship within the range of 3.16 × 102 − 3.16 × 106 copies/µL.

Standard curve for TaqMan qPCR of pMD19-INFB.

(A) Sensitivity of qPCR HPS detection; (B) Sensitivity of normal PCR detection; (C) Sensitivity of HPS qPCR established in this study for detection of viral nucleic acids; (D) Sensitivity of commercial kits for detection of viral nucleic acids. NC: Negative control; M: Marker with molecular weight 2000; All numbers in A-D indicate the number of dilutions.
Sensitivity assay for HPS qPCR
The qPCR amplification of the diluted DNA from positive plasmid demonstrated that the HPS detection system developed in this study could detect as low as 3.16 copies/µL (Fig. 5A). In contrast, conventional PCR only displayed weak bands in the electrophoresis channel when the plasmid concentration reached 3.16 × 103 copies/µL (Fig. 5B). This indicates that the system developed in this study is 1000 times more sensitive than conventional PCR. Furthermore, the system was able to specifically detect vaccine nucleic acid at a 106-fold dilution (Fig. 5C), whereas the competitor reagent could only detect it at a 104-fold dilution (Fig. 5D). Overall, the method established in this study shows a analytical sensitivity that is 100 times higher than that of commercial kits. It is important to note that while high analytical sensitivity contributes to improved diagnostic sensitivity, it does not necessarily equate to diagnostic sensitivity itself.
Precision experiment for HPS qPCR
Three different concentrations of pMD19-INFB DNA were chosen for testing, and the optimized systems were utilized for both intra-batch and inter-batch repeated experiments. As shown in Table 4, The coefficient of variation for each group in repeated experiments was less than 1%, indicating that the HPS qPCR method developed in this study demonstrates excellent repeatability.
Results of HPS qPCR interference experiments
The stability of the established HPS qPCR detection system was confirmed by testing with various types of endogenous and exogenous interfering substances. Table 5 indicates that the presence of feces, mucin, and fecal matter in the test samples slightly inhibited the detection efficacy of HPS. However, no interfering substances resulted in severe inhibitory effects. These findings demonstrate that the HPS qPCR detection system developed in this study exhibits strong resistance to a range of endogenous and exogenous interfering substances.
Clinical sample testing
In this study, 248 samples were tested using both the method developed here and commercially available kits. As shown in Table 6, Using national standards as the benchmark, the probability of a sample being identified as positive by the control kit was 6.05% (95% CI 4.08%−8.02%), while the probability of a sample being identified as positive by our method was 9.27% (95% CI 7.07%−11.47%). the positive and negative percent agreement indicates that the detection method developed in this study performs comparably to the national standard. Samples that tested positive with the experimental system but negative with the competitor’s kit were retested to confirm the positive results (Fig. 6). Furthermore, with the national standard as the reference and Cohen’s Kappa as the evaluation index, the κ value of the control kit was 0.776, while the κ value of our study was 1. These results clearly demonstrate that the qPCR detection system developed in this study has a higher detection rate compared to the competitor kits.

HPS sample retest results.
Discussion
HPS is commonly found in the environment. This slow-growing bacterium has specific nutritional requirements and is highly sensitive to fluctuations in its surrounding environment34,35. Consequently, any alterations in culture conditions during the collection and transportation of HPS samples to the laboratory may lead to a reduction in HPS survival. This difficulty further limits the use of bacterial isolation and culture as the ‘gold standard’ for diagnosing HPS36,37,38. Pigs are frequently co-infected with HPS and other bacteria and viruses. In instances of co-infection, the mortality rate among pigs affected by HPS is significantly elevated39,40. Furthermore, the weak cross-protection observed between HPS serotypes considerably diminishes the efficacy of vaccines12,41, thereby increasing the likelihood of large-scale epidemic outbreaks. Consequently, regardless of vaccination status, timely and regular detection of HPS is essential, as it can effectively reduce the economic losses associated with pigs infected by HPS.
Currently, the mainstream methods used for HPS detection are categorized into serological and molecular biology techniques. Due to the high cost associated with individual tests and the challenges in detecting early infections using serological diagnostic methods, molecular biology approaches are frequently employed in practical applications. Among these, qPCR technology offers advantages in terms of a mature technical system and high detection sensitivity when compared to conventional PCR, loop-mediated isothermal amplification, and nucleic acid molecular hybridization. In conventional PCR, the 16 S rRNAgene is commonly utilized as a target for primer design, serving as a taxonomic and phylogenetic marker35,42. However, in qPCR diagnostics, no significantly distinct regions within the 16 S rRNA gene have been identified between the primer sequences of HPS and other species, making it difficult to differentiate H. parasuisfrom closely related species43. Research conducted by C. Turni et al. (2010). has indicated that IFNB serves as a reliable surrogate target for 16 S rRNA, effectively distinguishing H. parasuisfrom all other closely related species35. Currently, numerous qPCR diagnostic methods targeting the IFNBgene for HPS have been developed44,45,46. This study has also designed detection primers and probes based on the conserved region of the housekeeping INFB gene across 15 serotypes of HPS to facilitate HPS detection.
To prove the resistance of the established HPS qPCR to these interfering substances, we mixed several potential interferents with the amplified nucleic acid samples for experimentation, a consideration that was absent in previous HPS qPCR studies. The results indicate that the qPCR detection system developed in this study exhibits strong anti-interference capability. Among all tested substances, feces, mucin (from semen), and milk were found to inhibit the detection efficacy of HPS. This may be attributed to the high concentrations of these interfering substances, which contain impurities and nucleic acids that are challenging to separate. Furthermore, aerosol contamination is a primary contributor to false positive test results47,48. In this study, we incorporated a UNG enzyme contamination prevention system into the qPCR amplification framework, effectively eliminating false positives caused by nucleic acid amplicon aerosols.
This study has several limitations. Although we verified the specificity of the primer probes using bioinformatics methods during the primer design process, we were unable to obtain species for comparison with HPS. Consequently, during the specificity detection phase, only common pig pathogens were utilized for specific detection. Furthermore, in the HPS qPCR process, HPS strains were not isolated from the samples and cultured for identification, which constitutes another limitation of this study. However, subsequent identification of real samples may partially address this limitation.
Conclusion
The study utilized the INFB gene of HPS for primer probe design and optimized the primer concentration, probe concentration, and annealing temperature in the reaction system through primer screening. The optimal reaction conditions for HPS qPCR were determined as follows: using F1/R1 as the primer, a primer concentration of 0.3 µL, a probe concentration of 0.2 µL, and an annealing temperature of 60℃. The established detection system specifically amplified HPS among common pig pathogens, achieving a detection sensitivity of less than 10 copies/µL. The coefficient of variation (CV) values for both intra-batch and inter-batch repeatability experiments were below 1%. Additionally, the system demonstrated good resistance to 8 endogenous and 6 exogenous interfering substances. Ultimately, the positive detection rate of HPS in real sample testing surpassed that of competing kits.
Data availability
The datasets used and analysed during the current study available from the corresponding author on reasonable request.
References
Fu, S. et al. Baicalin inhibits Haemophilus Parasuis-Induced High-Mobility group box 1 release during inflammation. Int. J. Mol. Sci. 19(5) (2018).
Feng, S. et al. Either fadD1 or fadD2, which encode acyl-CoA synthetase, is essential for the survival of Haemophilus parasuis SC096. Front. Cell. Infect. Microbiol. 7, 72 (2017).
Dickerman, A., Bandara, A. B. & Inzana, T. J. Phylogenomic analysis of Haemophilus parasuis and proposed reclassification to glaesserella Parasuis, gen. Nov., comb. Nov. Int. J. Syst. Evol. MicroBiol. 70 (1), 180–186 (2020).
Zhang, B. et al. Transcriptome analysis of heat resistance regulated by quorum sensing system in glaesserella parasuis. Front. Microbiol. 13, 968460 (2022).
Yue, C. et al. Autophagy is a defense mechanism inhibiting invasion and inflammation during High-Virulent Haemophilus parasuis infection in PK-15 cells, frontiers in cellular and infection microbiology 9 93. (2019).
Wang, L. D. C. Diagnosis and treatment of Haemophilus Parasuis in pigs, 20 91–93 (2022). (in Chinese).
Huang, A. et al. Dosing regimen of aditoprim and sulfamethoxazole combination for the glaesserella parasuis containing resistance and virulence genes. Pharmaceutics 14(10) (2022).
Li, J. et al. Secondary Haemophilus parasuis infection enhances highly pathogenic Porcine reproductive and respiratory syndrome virus (HP-PRRSV) infection-mediated inflammatory responses. Vet. Microbiol. 204, 35–42 (2017).
Liu, S. et al. Coinfection with Haemophilus parasuis serovar 4 increases the virulence of Porcine circovirus type 2 in piglets. Virol. J. 14 (1), 227 (2017).
Zhao, Z. et al. The occurrence of Bordetella bronchiseptica in pigs with clinical respiratory disease. Veterinary J. (London England: 1997). 188 (3), 337–340 (2011).
Hua, K. et al. Haemophilus parasuis infection disrupts adherens junctions and initializes EMT dependent on canonical Wnt/β-Catenin signaling pathway. Front. Cell. Infect. Microbiol. 8, 324 (2018).
Huang, J. et al. Pigs overexpressing Porcine β-Defensin 2 display increased resilience to glaesserella parasuis infection. Antibiot. (Basel Switzerland) 9(12) (2020).
ZhouQ. et al. Two glycosyltransferase genes of Haemophilus parasuis SC096 implicated in lipooligosaccharide biosynthesis, serum resistance, adherence, and invasion. Front. Cell. Infect. Microbiol. 6, 100 (2016).
Fu, S. et al. Transcriptional profiling of host cell responses to virulent Haemophilus Parasuis: new insights into pathogenesis. Int. J. Mol. Sci. 19(5) (2018).
Zhang, P. et al. Comparison of de Novo assembly strategies for bacterial genomes. Int. J. Mol. Sci. 22(14) (2021).
Cai, X. et al. Serological characterization of Haemophilus parasuis isolates from China. Vet. Microbiol. 111 (3–4), 231–236 (2005).
Zhou, X. et al. Distribution of antimicrobial resistance among different serovars of Haemophilus parasuis isolates. Vet. Microbiol. 141 (1–2), 168–173 (2010).
Zhang, Y., Li, Y., Yuan, W., Xia, Y. & Shen, Y. Autophagy is associated with pathogenesis of Haemophilus parasuis. Front. Microbiol. 7, 1423 (2016).
Jiang, C. et al. Effect of cAMP receptor protein gene on growth characteristics and stress resistance of Haemophilus parasuis serovar 5, frontiers in cellular and infection microbiology 10 19. (2020).
Gong, H. et al. Pleural thickening induced by glaesserella parasuis infection was linked to increased collagen and Elastin. Front. Cell. Infect. Microbiol. 12, 952377 (2022).
Macedo, N. et al. Molecular characterization of glaesserella parasuis strains isolated from North America, Europe and Asia by serotyping PCR and LS-PCR. Vet. Res. 52 (1), 68 (2021).
Matiašková, K. et al. The role of antibodies against the crude capsular extract in the immune response of Porcine alveolar macrophages to in vitro infection of various serovars of glaesserella (Haemophilus) parasuis. Front. Immunol. 12, 635097 (2021).
Cui, Y. et al. Ct value-based real time PCR serotyping of glaesserella parasuis. Vet. Microbiol. 254, 109011 (2021).
Macedo, N., Oliveira, S., Torremorell, M. & Rovira, A. Immune response to oligopeptide permease A (OppA) protein in pigs naturally and experimentally infected with Haemophilus parasuis. Res. Vet. Sci. 107, 62–67 (2016).
Miniats, O. P., Smart, N. L. & Ewert, E. Vaccination of gnotobiotic primary specific pathogen-free pigs against Haemophilus parasuis. Can. J. Veterinary Res. = Revue Canadienne De Recherche Veterinaire. 55 (1), 33–36 (1991).
Miniats, O. P., Smart, N. L. & Rosendal, S. Cross protection among Haemophilus parasuis strains in immunized gnotobiotic pigs. Can. J. Veterinary Res. = Revue Canadienne De Recherche Veterinaire. 55 (1), 37–41 (1991).
Takahashi, K. et al. A cross-protection experiment in pigs vaccinated with Haemophilus parasuis serovars 2 and 5 bacterins, and evaluation of a bivalent vaccine under laboratory and field conditions. J. Veterinary Med. Sci. 63 (5), 487–491 (2001).
Oliveira, S., Galina, L. & Pijoan, C. Development of a PCR test to diagnose Haemophilus parasuis infections. J. Veterinary Diagn. Investigation: Official Publication Am. Association Veterinary Lab. Diagnosticians Inc. 13 (6), 495–501 (2001).
Buckwalter, S. P. et al. Inhibition controls for qualitative real-time PCR assays: are they necessary for all specimen matrices? J. Clin. Microbiol. 52 (6), 2139–2143 (2014).
Acharya, K. R., Dhand, N. K., Whittington, R. J. & Plain, K. M. PCR Inhibition of a quantitative PCR for detection of Mycobacterium avium subspecies paratuberculosis DNA in feces: diagnostic implications and potential solutions. Front. Microbiol. 8, 115 (2017).
Konduru, B., Sagi, S. & Parida, M. Dry reagent-based Multiplex real-time PCR Assays for Specific Identification of Chicken, Mutton, Beef and Pork in Raw and Processed Meat Products (European Food Research and Technology, 2021).
Song, L. et al. Mucosal and systemic immune responses to influenza H7N9 antigen HA1-2 Co-Delivered intranasally with Flagellin or polyethyleneimine in mice and chickens. Front. Immunol. 8, 326 (2017).
Dong, X. et al. Draft genome of the honey bee ectoparasitic mite, tropilaelaps Mercedesae, is shaped by the parasitic life history. GigaScience 6 (3), 1–17 (2017).
Morozumi, T. & Hiramune, T. Effect of temperature on the survival of Haemophilus parasuis in physiological saline. Natl. Inst. Anim. Health Q. 22 (2), 90–91 (1982).
Turni, C., Pyke, M. & Blackall, P. J. Validation of a real-time PCR for Haemophilus parasuis. J. Appl. Microbiol. 108 (4), 1323–1331 (2010).
Sadamoto, S. et al. Comparison approach for identifying missed invasive fungal infections in Formalin-Fixed, Paraffin-Embedded autopsy specimens. J. fungi (Basel Switzerland) 8(4) (2022).
Nuutila, J., Hohenthal, U., Oksi, J. & Jalava-Karvinen, P. Rapid detection of bacterial infection using a novel single-tube, four-colour flow cytometric method: comparison with PCT and CRP. EBioMedicine 74, 103724 (2021).
Yu, M. et al. A visual assay of a Loop-Mediated isothermal amplification based vertical immunoassay for SARS-CoV-2 RNA detection. Front. Microbiol. 13, 932698 (2022).
Hričínová, M., Holoda, E., Mudroňová, D. & Ondrašovičová, S. Multiplex PCR assay for detection of Actinobacillus pleuropneumoniae, Pasteurella multocida and Haemophilus parasuis in lungs of pigs from a slaughterhouse. Folia Microbiol. 55 (6), 635–640 (2010).
Kavanová, L. et al. Concurrent infection with Porcine reproductive and respiratory syndrome virus and Haemophilus parasuis in two types of Porcine macrophages: apoptosis, production of ROS and formation of multinucleated giant cells. Vet. Res. 48 (1), 28 (2017).
Nielsen, R. Pathogenicity and immunity studies of Haemophilus parasuis serotypes. Acta Vet. Scand. 34 (2), 193–198 (1993).
Angen, O., Oliveira, S., Ahrens, P., Svensmark, B. & Leser, T. D. Development of an improved species specific PCR test for detection of Haemophilus parasuis. Vet. Microbiol. 119 (2–4), 266–276 (2007).
Hedegaard, J. et al. Investigation of the translation-initiation factor IF2 gene, InfB, as a tool to study the population structure of Streptococcus agalactiae, microbiology (Reading, England) 146 (Pt 7) 1661–1670. (2000).
Turni, C., Pyke, M. & Blackall, P. J. Validation of a real-time PCR for Haemophilus parasuis. J. Appl. Microbiol. 108(4) (2010).
Zhao, W. H., Luo, Q. B., Xin, P., Chi, C. F. & Kun-Lai, S. Preparation, Identification, and Activity Evaluation of Ten Antioxidant Peptides from Protein Hydrolysate of Swim Bladders of Miiuy Croaker (Miichthys Miiuy) (JOURNAL OF FUNCTIONAL FOODS, 2018).
Frandoloso, R., Martínez-Martínez, S., Rodríguez‐Ferri, E. F. & Gutiérrez‐Martín, C. B. Comparison of real-time PCR and culture isolation in colostrum-deprived pigs immunized and challenged with Haemophilus parasuis. Lett. Appl. Microbiol. 54 (2), 149–152 (2012).
Chen, Y. et al. Contamination-free visual detection of SARS-CoV-2 with CRISPR/Cas12a: A promising method in the point-of-care detection. Biosens. Bioelectronics: Int. J. Prof. Involved Res. Technol. Appl. Biosensers Relat. Devices. 169-, 169 (2020).
Sun, Y., Yu, L., Liu, C., Ye, S. & Huang, W. One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J. Translational Med. 19(1) (2021).
Funding
This work was financially supported by Jilin Province Science and Technology Development Plan Project (20210204115YY, YDZJ202201ZYTS662).
Author information
Authors and Affiliations
Contributions
H.J. and Y.X: investigation, methodology, data curation, formal analysis, conceptualization, writing—original draft. J.W.: investigation, methodology, conceptualization, data curation, formal analysis, and writing— original draft. Z.W.: investigation, data curation, methodology, and writing—original draft. S.W.: investigation, methodology, formal analysis, and data curation. C.D.: investigation, methodology. Y.Z.: investigation, formal analysis. Y.S. and C.K.: project administration, supervision, and writing—review and editing. All the authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, H., Xiao, X., Wang, J. et al. Establishment and validation of a simple and accurate qPCR detection method for Haemophilus parasuis. Sci Rep 15, 8264 (2025). https://doi.org/10.1038/s41598-025-92803-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-92803-1
