
Abstract
This study aims to explore how exercise enhances mitochondrial regulation and mitigates pathological cardiac hypertrophy. Rat groups were assigned as the control group (CN, n = 8), sham group (sham, n = 8), model group (SC, n = 16) and exercise group (SE, n = 20). A bioinformatics analysis was conducted to uncover the underlying mechanisms.H9C2 cells were divided into: the Ang II 0 h group (CON), Ang II 48 h group (Ang II), Ang II 48 h + sh-control group (sh-GFP + Ang II), Ang II 48 h + sh-ndufb10 group (sh-ndufb10 + Ang II), Ang II 48 h + overexpressedndufb10 control group (Ad-GFP + Ang II) and Ang II 48 h + over-expressedndufb10group (Ad-ndufb10 + Ang II). Mitochondrial function was measured. mRNA and protein expression were assessed by qPCR or western blot analysis respectively. In the SC group, a significant increase was observed in cardiomyocyte diameter, cardiac function, autophagy, and apoptosis. After 8 weeks of swimming exercise, there was a substantial reduction in cardiomyocyte diameter, an improvement in cardiac function, a mitigation of mitochondrial fission and autophagy. Ndufb10 was markedly enriched in oxidative phosphorylation and downregulated in the SC group, while it was upregulated in the SE group. In the sh-ndufb10 group, mitochondrial fusion was suppressed; fission and autophagy were further facilitated; mitochondrial membrane potential, mPTP, and ROS levels increased; and TUNEL positive nuclei and apoptosis-related proteins showed significant upregulation. Overexpression of ndufb10 reversed pathological hypertrophy, mitochondrial autophagy, mitochondrial dysfunction, and cardiomyocyte apoptosis in vitro. Swimming exercise improves mitochondrial abnormalities and reduces cardiomyocyte hypertrophy through regulation of the ndufb10 in left ventricular hypertrophy.
Similar content being viewed by others

Introduction
Myocardial hypertrophy is a complex series of changes that precede heart failure (HF) and are accompanied by increased fibrosis, ventricular dysfunction, mitochondrial dysfunction and abnormal mitochondrial autophagy in response to various stimulating factors1. Mitochondria play an essential role in maintaining the normal energy supply of the heart. Under normal physiological conditions, the mitochondrial network controls its own size and morphology through continuous fusion and division, directly affecting oxidative phosphorylation2,3. Mitochondrial dysfunction disrupts the function of the mitochondrial electron respiratory chain and the balance of mitochondrial dynamics (fusion and division), ultimately resulting in an imbalance in mitochondrial homeostasis4.
Mitochondrial abnormalities, cardiomyocyte dysfunction, and cardiac fibrosis contribute to pathological cardiac hypertrophy and heart failure5,6. Impaired mitochondria may accelerate the production of reactive oxygen species (ROS) and cytochrome-C, further contributing to cardiomyocyte injury and programmed cell death, ultimately leading to pathological cardiac hypertrophy and heart failure7,8. During cellular development, cells can either survive through the inhibition of apoptosis by autophagy or undergo cell death caused by autophagy itself or occasionally through the interaction of autophagy with apoptosis, suggesting a potential structural correlation between autophagy and apoptosis9,10. In addition, mechanical changes in mitochondrial dynamics (fusion fission homeostasis imbalance), mitochondrial fragmentation, mitochondrial morphological and quantitative abnormalities, and energy metabolism disorders occur during many pathological processes, which disrupt mitochondrial homeostasis, impair cardiac bioenergy and excessive oxidative stress, and eventually induce left ventricular hypertrophy (LVH)2,11. The NDUF family, involved in the assembly of complex I, is a significant auxiliary subunit. Reduced expression of NDUF family proteins has been closely associated with complex I dysfunction in various tissues12, impacting the myocardial energy supply and the heart’s ability to pump blood continuously13,14. However, the specific mechanism of the NDUF family in pathological myocardial hypertrophy requires further investigation.
Exercise regulates myocardial mitochondrial homeostasis, promoting the removal of damaged mitochondria and improving the function and quality of mitochondria in cardiomyocytes15, which are mediated by the modulation of autophagy pathways, including the PTEN-induced putative kinase 1 (PINK1)/Parkin and apoptosis pathways NIX/BNIP316,17. Additionally, exercise facilitates mitochondrial biogenesis and enhances mitochondrial oxidative respiration, resulting in decreased ROS production18,19 and the restoration of mitochondrial homeostasis. However, the molecular mechanism underlying these effects of exercise remains to be fully elucidated.
In this study, we hypothesized that exercise may contribute to the alleviation of TAC-induced pressure overload LVH and cardiac dysfunction via mitochondrial autophagy and cardiomyocyte, and that the underlying mechanisms and signalling pathways can be further delineated. The findings from our study will contribute to the development of novel and noninvasive strategies for cardioprotection against cardiovascular remodelling-induced dysfunction involved in human diseases.
Results
Effect of 8-week swimming exercise on TAC-induced ventricular dysfunction in the left ventricle
52 rats participated in the experiment, and 6 rats died after TAC, with 4 deaths attributed to intestinal obstruction, resulting in a mortality rate of 11.5%, no rats died in sham group. Additionally, during swimming, 4 rats died, with 1 death attributed to stress reaction, yielding a swimming mortality rate of 8.7%.
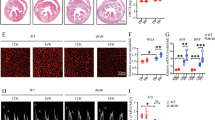
The results of various parameters, including LVSd, LVIDd, LVPWd, LVPWs, LVSs, LVIDs, EF, and FS, are summarized in Table 1. In the SC group, there was a significant increase in LVSd (1. 5 ± 0.06 vs. 1.19 ± 0.04, P < 0.05), LVPWd (2.10 ± 0.10 vs. 1.45 ± 0.14, P < 0.05), LVPWs (2.97 ± 0.07 vs. 2.34 ± 0.07, P < 0.05), LVIDd (7.93 ± 0.09 vs. 6.66 ± 0.29, P < 0.05), and LVIDs (5.13 ± 0.09 vs. 3.54 ± 0.21, P < 0.05) (Fig. 1A–G), accompanied by a decrease in EF (0.72 ± 0.02 vs. 0.83 ± 0.01, P < 0.05) (Fig. 1H) and FS (0.39 ± 0.01 vs. 0.47 ± 0.01, P < 0.05) (Fig. 1I), when compared to the CN rats. Intriguingly, the 8-week swimming exercise regimen in the SE group exhibited a reversal of the elevated left ventricular end-systolic dimension (LVSd) (1.300 ± 0.00 vs. 1.19 ± 0.04, P > 0.05), left ventricular posterior wall thickness at end-diastole (LVPWd) (1.33 ± 0.03 vs. 1.45 ± 0.14, P > 0.05), left ventricular posterior wall thickness at end-systole (LVPWs) (2.31 ± 0.06 vs. 2.34 ± 0.07, P > 0.05), and left ventricular internal dimension at end-systole (LVIDs) (2.34 ± 0.07 vs. 3.54 ± 0.21, P > 0.05), which became nearly the same as those of the CN group. Moreover, the exercise regimen improved the decreased ejection fraction (EF) (0.77 ± 0.01 vs. 0.72 ± 0.02, P < 0.05) in the SE group compared to the SC group. These findings suggest that exercise may alleviate TAC-induced cardiomyocyte hypertrophy and ventricular dysfunction.


Exercise alleviated TAC-induced ventricular dysfunction and pathological remodelling in rats. Diastolic and systolic function parameters in rats (A–I) were calculated through echocardiography, n = 3. (A) Representative dropper images, (B) LVSd: Left ventricular end-diastolic septum thickness, (C) LVIDd: Left ventricular end-diastolic diameter (D) LVPWd: Left ventricular end-diastolic posterior wall thickness, (E) LVPWs: Left ventricular end-systolic posterior wall thickness, (F) LVSs: Left ventricular end-systolic septum thickness, (G) LVIDs: Left ventricular end-systolic diameter, (H) EF: ejection fraction, (I) FS: Shortening fraction; (J) Representative heart sections examined by haematoxylin and eosin (H&E) staining (upper), Masson staining of myocardial fibrosis (lower). Scale bar, 50 mm, representative qPCR analysis (K) and western blot (original blots/gels are presented in Supplementary 2 Fig. 1). (L) Representative ANP and BNP expression levels in hearts, n = 4, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
Additionally, as demonstrated in Table 2, the heart weight to body weight ratio (HW/BW) significantly increased in the SC group (4.30 ± 0.34) compared to the CN group (3.29 ± 0.26, P < 0.05). Similarly, the left ventricle weight to body weight ratio (LV/BW) was significantly higher in the SC group (3.06 ± 0.22) than in the CN group (2.14 ± 0.23, P < 0.05). In contrast, in the SE group, HW/BW (4.13 ± 0.24) and LV/BW (2.72 ± 0.23) both decreased compared to the SC group (P < 0.05). Notably, there was no statistically significant difference between the CN and sham rats, indicating that the surgical procedure had no impact on left ventricular hypertrophy (LVH).
Beneficial effects of 8-week swimming exercise on TAC-induced left ventricular pathological remodelling
To assess pathological myocardial hypertrophy in the SC group and the protective impact of exercise in the SE group, we stained the left ventricular myocardial tissue with HE and Masson stains, followed by quantification of the myocardial fibrosis area using ImageJ software. The cross-sectional myocyte area and collagen deposition were significantly greater in the SC group than in the CN group (Fig. 1J–K). However, these indicators markedly decreased after 8 weeks of exercise in the SE group compared to the SC group. Concurrent with the reduction in heart size and cardiac fibrosis, the analysis of the LV myocardium samples from SE rats compared to those from SC rats via real-time quantitative polymerase chain reaction (qPCR) (Fig. 1L) and western blot (Fig. 1M) revealed significantly lower expression levels of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) after exercise. Importantly, surgical intervention had no statistically significant effect on our results, as evidenced by the absence of differences between the CN and sham rats.
Exercise mitigates TAC-induced mitochondrial autophagy
To assess the impact of 8 weeks of swimming exercise on mitochondrial function, we conducted a transmission electron microscopy (TEM) assay. TEM analysis of rat heart sections revealed several noteworthy findings. Compared with the CN group, the SC group showed an increase in fragmented mitochondria, along with a significant rise in autophagic lysosomes. Additionally, severe mitochondrial oedema and vacuolization were observed in the SC group compared to both CN groups. However, compared to the SC group, in the SE group, we observed a reduction in mitochondrial fission, oedema, and autophagic lysosomes (Fig. 2A). In line with the observed disruption in mitochondrial dynamics in the TEM images, we found that the expression levels of the mitochondrial fusion protein Mitofusin1 (MFN1) decreased in the SC group compared with the CN group but were reversed in the SE group compared with the SC group (Fig. 2C). Furthermore, markers related to mitochondrial autophagy, such as microtubule-associated protein 1 light chain 3 alpha (LC3II), the Parkin pathway, and the NIX/BNIP3 pathway, were upregulated in the SC group compared to the CN group. Exercise in the SE group appeared to mitigate excessive autophagy (Fig. 2B,C). Notably, there were no significant differences observed between the CN and sham groups, indicating that the surgical procedure had no discernible effect on mitochondrial autophagy.

Exercise reduced TAC-induced mitochondrial autophagy (A) Representative TEM images of rat LV cross sections, n = 2. Representative qPCR analysis of mitophagy (B), n = 4. (C) Representative western blot (original blots/gels are presented in Supplementary 2 Fig. 2–6) analysis and quantification of proteins involved in mitophagy, n = 4, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
Exercise attenuates TAC-induced cardiomyocyte apoptosis
To further investigate the occurrence of myocardial apoptosis, we conducted TUNEL assays on rats. The staining images and quantification of TUNEL-positive nuclei revealed a significant increase in cardiomyocyte in the SC group compared to the CN group. Notably, swimming exercise reversed cardiomyocyte in the SE group compared to the SC group (Fig. 3A). Consistent with these findings, both the mRNA and protein expression levels of apoptosis factors, such as caspase-3 and caspase-9, were significantly upregulated in the SC group compared to the CN group and downregulated in the SE group compared to the SC group (Fig. 3B,C). Cardiomyocyte is also supported by the decreased expression of BCL-2 and increased expression of BAX in the pathological conditions of the SC group compared to the CN group. Importantly, swimming exercise alleviated the activation of apoptosis in the SE group compared to the SC group (Fig. 3B,C).

TUNEL assay of cardiac myocyte apoptosis in the hearts. (A) Quantification of TUNEL-positive nuclei n = 3. Representative qPCR (B) and western blot (C) analysis (original blots/gels are presented in Supplementary 2 Fig. 7–9) of apoptosis-related mRNA and protein, n = 3, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
RNA expression analysis via RNA-Seq
To explore the underlying molecular mechanisms, we conducted RNA-Seq on four rats from both the SC and SE groups. This analysis revealed 161 upregulated and 116 downregulated genes involved in various biological processes when comparing the SC and SE groups (Fig. 4A–C). KEGG analysis indicated an upregulation of the oxidative phosphorylation pathway (Fig. 4D). Within this pathway, 13 genes (gene set 1) were significantly enriched (Fig. 4D), and a heatmap displayed their differential expression levels between the SC and SE groups (Fig. 4E). Our study identified 135 interaction genes (gene set 2) related to gene set 1, and 10 key drive genes (KDAs) were determined (Fig. 4F).Notably, a highlighted ndufb10 as the sole key gene driving the oxidative phosphorylation pathway (Fig. 4G). Subsequently, we conducted qPCR and western blot analyses of ndufb10 expression in the SC and SE groups, and the results were consistent with the RNA-Seq data (Fig. 4H). These findings suggest that exercise may improve LVH, potentially by regulating ndufb10.


Exercise improves LVH by regulating Ndufb10. RNA-sequencing analysis of the left ventricles of SC and SE rats, n = 4. (A) Violin plot representing distribution of FPKM values; the five characteristic values indicate maximum, upper quartile, mid-value, lower quartile, and minimum, respectively; (B) Volcano map of DEGs, (C) Enriched GO terms of biological process, cellular component and molecular function; (D) KEGG pathway enrichment chart; (E) Heatmap of genes enriched in oxidative phosphorylation pathway; (F) PPI network including 10 KDAs (red) and 20 extended genes (pink); (G) Venn diagram of gene set 1 and KDA genes. (H) qPCR and western blot analysis (original blots/gels are presented in Supplementary 2 Fig. 10) of ndufb10 expression, n = 4. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
In vitro study of pathological cardiac hypertrophy
To delve deeper into the role of ndufb10 in pathological cardiac hypertrophy, we utilized an Angiotensin II (Ang II)-induced H9C2 cell model. H9C2 cells were collected 48 h after stimulation with Ang II at a concentration of 100 nmol/L. The evaluation involved both inverted microscope images (Supplementary Fig. 2A) and qPCR analysis (Supplementary Fig. 2B), which revealed an increase in cell diameter and elevated expression of anp and bnp in the Ang II group compared to the CON group. These findings confirm the successful establishment of a hypertrophic cell model.
In vitro Ndufb10 knockdown model
To gain further insights into the role of ndufb10 in LVH, we established an in vitro ndufb10 knockdown model. Fluorescence images (Fig. 5A) and qPCR analysis of ndufb10 expression (Fig. 5B) demonstrated a transfection efficiency of 70% and a significant reduction in ndufb10 mRNA expression, confirming the successful transfection of sh-ndufb10 and sh-GFP adenoviruses. After sh-ndufb10 infection, H9C2 cells were incubated with Ang II for 48 h to simulate pathological hypertrophy in vitro. The mRNA expression of bnp was not significantly different between the Ang II group and the sh-GFP + Ang II group (Fig. 5C). Notably, bnp mRNA expression was further elevated in the sh-ndufb10 + Ang II group compared to the Ang II group (Fig. 5C). In Fig. 5D, we observed a marked increase in the protein expression of DRP1, a mitochondrial fusion marker, and a significant downregulation in the protein expression of OPA1, a fusion protein marker, in the sh-ndufb10 + Ang II group compared to the Ang II group. Additionally, LC3 II was significantly increased in the Ang II and sh-ndufb10 + Ang II groups (Fig. 5E).

Knockdown of ndufb10 aggregates Ang II-induced mitochondrial dynamic abnormalities and autophagy. (A) Representative cell size and transfection efficiency of adenovirus. (B) ndufb10 expression in sh-GFP and sh-ndufb10 cells, n = 4. (C) bnp expression in cells, n = 4. (D) Expression levels of mitochondrial fission and fusion proteins, n = 4. (E) Expression levels of mitochondrial autophagy proteins, n = 4, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software, original blots/gels are presented in Supplementary 2 Fig. 11–16. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
Furthermore, the mitochondrial autophagy signalling pathways PARKIN and NIX/BNIP3 were activated in both the Ang II group and the sh-ndufb10 + Ang II group (Fig. 5E). Moreover, mitochondrial dysfunction was aggravated in the sh-ndufb10 + Ang II group, as confirmed by increased mPTP opening (Fig. 6A, above), ROS production (Fig. 6A, below), decreased mitochondrial membrane potential (Fig. 6B), and NDUFB10 expression (Fig. 6C).


Knockdown of ndufb10 aggregates Ang II-induced mitochondrial dysfunction and apoptosis. (A) Representative mitochondrial mPTP opening (above) and fluorogram of ROS production (below) and their quantitative analysis, n = 3. (B) Mitochondrial membrane potential and its quantitative analysis, n = 3. (C) Expression level of the NDUFB10 protein. n = 3; (D) TUNEL assay of apoptosis in H9C2 cells. Quantification of TUNEL-positive nuclei, n = 3. (E) Expression levels of apoptosis-related proteins, n = 3, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software, original blots/gels are presented in Supplementary 2 Fig. 17–19. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
To assess cell apoptosis, cells in the four groups underwent TUNEL assays and western blot analysis. Quantification of TUNEL-positive nuclei increased by approximately 1.28-fold (Fig. 6D), and apoptosis-related proteins (CASPASE3, CASPASE9, and BAX) were notably upregulated (Fig. 6E) in the sh-ndufb10 + Ang II group compared to the Ang II group.
In vitro Ndufb10 overexpression model
To complement our previous observations regarding mitochondrial homeostasis and LVH, we investigated whether the expression of ndufb10 would influence mitochondrial morphology in H9C2 cardiac cells. We successfully established a ndufb10 overexpression model with a transfection efficiency of 70% (Fig. 7A), resulting in increased expression of ndufb10 mRNA (Fig. 7B). Subsequently, the cells were incubated with Ang II for 48 h following ndufb10 overexpression to simulate pathological hypertrophy in vitro.


Overexpression of ndufb10 ameliorated Ang II-induced mitochondrial dynamic abnormalities and autophagy. (A) Representative cell size and transfection efficiency of adenovirus. (B) ndufb10 expression in Ad-GFP and Ad-ndufb10 cells, n = 4. (C) bnp expression in cells, n = 4. (D) Expression levels of mitochondrial fission and fusion proteins, n = 4. (E) Expression levels of mitochondrial autophagy proteins, n = 4, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software, original blots/gels are presented in Supplementary 2 Fig. 20–23. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
As shown in Fig. 7C, the mRNA expression of bnp was significantly increased in both the Ang II and Ad-GFP + Ang II groups compared to the CON group, but this increase was reversed in the Ad-ndufb10 + Ang II group. Notably, there was no significant difference between the Ang II and Ad-GFP + Ang II groups, indicating that Ad-GFP had no effect on LVH.
The effects of ndufb10 expression on mitochondrial dynamics are evident in Fig. 7D, where the protein expression of FIS1 and DRP1 was significantly upregulated in both the Ang II and Ad-GFP + Ang II groups compared to the CON group. However, ndufb10 expression reversed the upregulation of these mitochondrial fission proteins. Moreover, the mitochondrial fusion protein APAF1 was decreased in both the Ang II and Ad-GFP + Ang II groups compared to the CON group, but it was improved in the Ad-ndufb10 + Ang II group.
Additionally, mitochondrial autophagy was triggered in both the Ang II and Ad-GFP + Ang II groups, as evidenced by the increased protein expression of LC3 II, PARKIN, and NIX/BNIP3, which was normalized with ndufb10 expression, except for PARKIN protein (Fig. 7E).
To further explore the effects of ndufb10 expression, we assessed mitochondrial function in cardiomyocytes. As shown in Fig. 8A, mitochondrial mPTP and ROS were significantly increased in both the Ang II and Ad-GFP + Ang II groups compared to the CON group, but this pathological condition was improved in the Ad-ndufb10 + Ang II group. Furthermore, mitochondrial membrane potential, as indicated by the green‒red fluorescence ratio and ndufb10 expression (Fig. 8B, C), decreased in both the Ang II and Ad-GFP + Ang II groups compared to the CON group but increased in the Ad-ndufb10 + Ang II group.


Overexpression of ndufb10 ameliorated Ang II-induced mitochondrial dysfunction and apoptosis. (A) Representative mitochondrial mPTP opening and its quantitative analysis, n = 3. (B) Mitochondrial membrane potential and its quantitative analysis, n = 3. (C) Expression level of ndufb10 mRNA. n = 3. (D) TUNEL assay of apoptosis in H9C2 cells. Quantification of TUNEL-positive nuclei, n = 3. (E) Expression levels of apoptosis-related proteins, n = 3, Chemiluminescence signals were visualized and cropped using Protein Simple Compass software, original blots/gels are presented in Supplementary 2 Fig. 24–25. The whole cell lysates were used for western blot. The data are presented as the mean ± SEM; *P < 0.05; **P < 0.01.
We also examined cell apoptosis using TUNEL assays, as shown in Fig. 8D. We observed a significant increase in apoptotic bodies in both the Ang II and Ad-GFP + Ang II groups, while the Ad-ndufb10 + Ang II group showed no difference compared to the CON group. Western blot analysis in Fig. 8E revealed a significant increase in the protein expression of CASPASE9, CYTOCHROME C, and BAX in both the Ang II and Ad-GFP + Ang II groups compared to the CON group. However, the Ad-ndufb10 + Ang II group reversed the expression of these proteins.
Discussion
Mitochondrial dysfunction is implicated in various cardiovascular diseases20, but the complex molecular mechanisms behind these alterations have remained elusive due to the intricate nature of mitochondria. Our study unveils a pivotal molecular event, which is the decreased expression of ndufb10 in left ventricular hypertrophy (LVH). The structured swimming exercise regimen effectively improved ventricular function and mitigated left ventricular hypertrophy (LVH). The loss of ndufb10 in H9C2 cells resulted in mitochondrial dysfunction and ultimately led to cardiomyocyte hypertrophy. Significantly, ndufb10 overexpression was effective in restoring mitochondrial function and ameliorating cardiomyocyte hypertrophy. Mitochondrial abnormalities are linked to several human diseases, including LVH13,20. Our findings not only elucidated the mitochondrial abnormalities triggered by LVH but also identify ndufb10 as a promising therapeutic target for managing LVH.
LVH is marked by cardiac enlargement and compromised ventricular function, frequently culminating in heart failure. In our study, we induced LVH in rats through TAC surgery, leading to significant ventricular dysfunction and notable increases inLVSd, LVIDd, LVPWd, LVPWs, and LVIDs.An 8-week swimming exercise significantly reduced left ventricle wall thickness (LVSd, LVPWd, LVPWs) and the LV/BW ratio while effectively mitigating left ventricular dysfunction (EF) in the SE group when compared to the SC group (Fig. 1). Notably, there was no significant difference in ventricular cavity size between the SC and SE groups. The main type of TAC-induced pathological cardiac hypertrophy is compensatory hypertrophy (concentric hypertrophy). As decreased blood supply, myocardial contractility enhanced and resulting in hyperplasia or hypertrophy of cardiomyocytes, which is consistent with our data (Fig. 1.) In our study, TAC surgery led to the upregulation of LV Wall thickness, including LVIDd, LVIDs, and FS, in the SC group (concentric hypertrophy). Contrary to LVSd, TAC surgery did not significantly reduce LVSs in the SC group, although it exhibited a similar trend compared to LVSd. This may be attributed to our TAC protocol, where the narrowing rate was only 50%, which might not have been strong enough to further increase LVSs. This partially explains the lack of statistical significance in LVSs between the SC and CN groups. As reported elsewhere21, exercise can also lead to cardiac hypertrophy characterized by increased ventricular chamber size (physiological eccentric hypertrophy). In the SE group, left ventricular hypertrophy (LVH) resulted from the combination of TAC and exercise stimuli. Previous evidence suggests that 8 weeks of swimming exercise can, to some extent, contribute to an increase in ventricular chamber dimensions22,23, which may explain why there were no statistically significant differences in LVIDd, LVIDs, and FS between the SC and SE groups (a mixed of eccentric and concentric hypertrophy). HE staining, aligned with echocardiography data, revealed a significant increase in cardiomyocyte size in the SC group. However, 8 weeks of swimming exercise effectively reduced this condition. Additionally, Masson staining indicated myocardial fibrosis proliferation in the SC group following TAC intervention, and swimming training also decreased cardiac fibrosis induced by TAC. These findings collectively highlight that TAC intervention in the SC group induced left ventricular hypertrophy (LVH), impaired ventricular function, and myocardial fibrosis proliferation, while 8 weeks of swimming exercise appears to mitigate these changes. This suggests that exercise can serve as a therapeutic approach to improve LVH and left ventricular dysfunction.
Autophagy is a fundamental process involved in maintaining cellular homeostasis and responding to pathological processes by degrading and recycling proteins and organelles through autophagic vesicles that fuse with lysosomes24. Previous research has explored the effects of exercise on regulating autophagy and apoptosis in LVH models. For instance, we previously observed increased autophagy activity and preserved apoptosis homeostasis following exercise in a rat model of pressure overload-induced LV dysfunction and remodelling21. In this study, we observed increased ruptured mitochondria and autophagosomes, along with decreased mitochondrial fusion marker (MFN1) expression and elevated mitochondrial autophagy markers (LC3 II, PARKIN, and BNIP3 expression) in TAC-induced LVH rats. These findings suggest that transitional autophagy, accumulated mitochondrial fragments, and autophagosomes (indicative of mitochondrial dynamics anomalies) closely parallel the pathological changes observed in the morphological and functional aspects of the left ventricle. Subsequently, we found that swimming exercise improved mitochondrial abnormalities, including abnormal autophagy, in response to TAC. These results indicate that TAC induces abnormalities in mitochondrial dynamics, and these abnormalities are ameliorated through exercise. In addition, this aligns with earlier studies suggesting that exercise can improve failing hearts by restoring cardiac autophagy flux25,26, indicating a close relationship between mitochondrial autophagy and cardiac remodelling and heart failure induced by TAC. Our observations contrast with those of Zhang et al.27, who reported impaired autophagy in pathological cardiac hypertrophy induced by ISO administration and decreased autophagy in diabetes and ageing-induced cardiac abnormalities28. However, methodological differences between these studies, such as the aetiological factors inducing pathological cardiac hypertrophy, may account for these discrepancies. It is conceivable that the degree and duration of pathological cardiac hypertrophy may be influenced by specific underlying causes. A surprising finding in our study was that the recruitment of PARKIN to mitochondria appeared to be independent of PINK1 after TAC. This suggests that cardiac mitophagy induced by haemodynamic stress may be regulated by unique signalling factors rather than relying solely on the well-known PINK1/PARKIN pathway. Our results extend previous findings, confirming that PINK1 is dispensable for mitochondrial recruitment of PARKIN and activation of mitophagy in cardiac myocytes29.
Furthermore, we observed improvements in cardiomyocyte triggered by TAC. BCL-2, a member of the same family as BAX, plays a role in regulating apoptosis activators by controlling mitochondrial membrane permeability. Our results showed a downregulation of BCL-2 and an upregulation of BAX in pathological conditions, which is consistent with other research indicating that BCL-2 binds to BAX, an apoptosis signal-activated molecule, forming heteromers that reduce membrane permeability30. These data suggest that swimming exercise effectively reverses cardiomyocyte apoptosis induced by mechanical stress. Non-myocytes apoptosis are the other cells except cardiomyocytes in the heart undergo apoptosis, including fibroblasts, endothelial cells, smooth muscle cells, and immune cells. In the process of pathological cardiac hypertrophy, both myocyte apoptosis and non- myocyte apoptosis exist simultaneously, which exacerbate myocardial hypertrophy via affecting the number, structure and function of cardiomyocytes, and ultimately lead to heart failure31. According our research, the Transmission Electron Microscopy (TEM) Assay shown that nucleus condensation, marginalization and nuclear enrichment occurred. Organelle mixing, cytoplasmic vacuolation and apoptotic corpuscles increased, which indicating non-myocyte possibly predominantly in TAC-induced cardiac hypertrophy32.
Mitochondrial respiratory complex I holds a pivotal role in mitochondrial function and metabolism. Mutations within mitochondrial complex I genes have been linked to cardiovascular diseases33. Ndufb10, a crucial auxiliary subunit of complex I, directly contributes to its assembly. Reductions in NDUF-encoded proteins have been associated with complex I dysfunction across various tissues12. While reported variants within the NDUF family/complex I have been linked to mitochondrial dysfunction and cardiac mitochondrial disorders in left ventricular hypertrophy (LVH)33, the underlying mechanisms remain unclear. In our study, we focused on the 161 upregulated genes in the SE group and identified that 13 of these genes were enriched in the oxidative phosphorylation pathway through analysis using Dr. Tom. Subsequently, we conducted a protein‒protein interaction (PPI) network analysis to predict protein‒protein interactions and identify genes, transcripts, or proteins that interact with these selected genes. This analysis led to the inclusion of 135 interaction genes (gene set 2) for further investigation. Using Dr. Tom and Cytoscape, we determined 10 key driver genes (KDAs), and a Venn diagram highlighted the prominent regulatory role of ndufb10 in the oxidative phosphorylation pathway. These findings align with those of previous research, supporting the notion that mutations in ndufb10 can lead to complex I deficiency, which is associated with fatal infantile lactic acidosis and cardiomyopathy12.
To further investigate the critical role of ndufb10 in improving mitochondria in LVH, we employed H9C2 cells with either gained or lost ndufb10 function. Ang II successfully induced LVH in cell models, as supported by the expression of anp and bnp and cell size. The data suggested that compared to the Ang II 48 h group, after 48 h of sh-ndufb10 infection, the cardiomyocyte pathological manifestations further deteriorated. We next examined mitochondrial morphology and found that ndufb10 knockdown augmented the protein levels of DRP1, LC3 II, PARKIN, NIX, and BNIP3 compared to those in the Ang II 48 h group. Meanwhile, the supressed P62 level was observed after Ang- II incubation and ndufb10 suppression, which hinting an activation of the autophagic process. The inverse relationship between LC3-II and P62 levels is consistent with an upregulation of autophagic flux, which facilitating autophagosome formation This enhanced autophagy may represent a compensatory mechanism in response to cellular stress induced by Ang-II. Autophagy is known to play a dual role in cardiomyocyte hypertrophy, it is able to either protect cells via degrading damaged organelles and proteins or contribute to pathology if dysregulated. While short-term activation of autophagy is probably cardioprotective, sustained autophagy could lead to cell death and exacerbate cardiac dysfunction34. This suggests that the loss of ndufb10 function exacerbates mitochondrial fission in the heart and disrupts the balance of autophagy flow, indicating mitochondrial dynamics abnormalities in the pathological state of left ventricular hypertrophy. Conversely, overexpression of ndufb10 reversed the disruption of mitochondrial fission and cardiac autophagic flow. However, ndufb10 expression did not affect PARKIN protein expression, indicating that mitochondrial autophagy activation likely occurs through the NIX/BNIP3 pathway instead of the PARKIN pathway and worsens with ndufb10 knockdown in myocardial tissue, while ndufb10 overexpression restrains excessive autophagy. Moreover, the data demonstrate that ndufb10 deficiency triggers mitochondrial oxidative stress and partially induces mitochondrial function disorder. In contrast, overexpression of ndufb10 improved mitochondrial homeostasis, as shown. These findings expand upon those of previous studies suggesting that oxidative stress, abnormal mitochondrial fragmentation accumulation, and dysregulated mitochondrial autophagy may synergistically drive cardiomyocyte death35,36. Other studies have demonstrated that disruption of mitochondrial homeostasis in the heart can lead to abnormal physiological processes, resulting in LVH, heart failure, and dilated cardiomyopathy37. Additionally, NDUF family members have been reported to regulate mitochondrial function by enhancing complex I activity and reducing ROS production14,38,39. In our study, ndufb10 knockdown also exacerbated apoptosis in the heart, as confirmed by the higher expression of CASPASE 3, CASPASE 9, and BAX proteins, while overexpression of ndufb10 reversed cardiomyocyte, indicating a link among apoptosis, mitochondrial abnormalities, and LVH. Our data rule out the possibility that ndufb10 improved mitochondrial homeostasis through the regulation of the NIX/BNIP3 pathways, which further alleviated Ang II-induced cardiomyocyte. This study suggests that exercise activates ndufb10, reduces the NIX/BNIP3 pathway to inhibit excessive mitochondrial autophagy, and mitigates excessive cardiomyocyte, ultimately ameliorating left ventricular hypertrophy (LVH). Similar trends have been observed in studies where exercise regulated the balance of mitochondrial fusion and fission, improved mitochondrial homeostasis, and reversed apoptosis caused by cardiac pathological states2,40,41,42. Notably, this is the first study, to our knowledge, to investigate the effectiveness of the complex I subunit ndufb10 in regulating mitochondrial homeostasis. However, it is important to acknowledge some limitations. While our hypotheses were supported statistically, further investigations are needed to determine whether knockout and overexpression of ndufb10 achieve the same effects in vivo.
Conclusions
8 weeks of swimming exercise resulted in a reduction in LVH. Simultaneously, abnormal mitochondrial autophagy, mitochondrial dysfunction, and cardiomyocyte were ameliorated. Furthermore, we observed the activation of the complex I subunit ndufb10 after 8 weeks of swimming exercise. The in vitro experiments demonstrated that knocking down ndufb10 exacerbated pathological hypertrophy, while overexpressing ndufb10 reversed it. In conclusion, 8 weeks of swimming exercise improves mitochondrial abnormalities and reduces cardiomyocyte by regulating the NIX/BNIP3 pathway through ndufb10 in the context of LVH.
Methods
All methods were performed in accordance with the relevant guidelines and regulations.
Animals housing and groups
Seven-week-old Sprague‒Dawley female rats (n = 52) weighing 200 ± 20 g were obtained from the Experimental Animal Center of the Three Gorges University (Certificate No. 42010200007182). All animal experimental protocols were approved by the Animal Care and Use Committees of Wuhan Sports University (protocol number 0087-202105-0101). All experiments were performed in accordance with relevant named guidelines and regulations All methods were carried out in accordance with ARRIVE guidelines and the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020). The rats were housed at a constant temperature of 21 ~ 24 °C, with a humidity of 50 ~ 60% and a 12-h light–dark cycle. After 1 week of adaptation, the rats were randomly assigned to four groups: the control group (CN, n = 8), sham group (sham, n = 8), model group (SC, n = 16) and exercise group (SE, n = 20).
LVH models and exercise protocol
As described previously21, LVH was induced by transverse aortic constriction (TAC) in the SE group following a 7-day adaptive feeding period for the animals. Briefly, under anaesthetized conditions, the abdomens of the rats were surgically opened in the SC and SE groups. The abdominal aorta was fully exposed and ligated using a 3-gauge needle, resulting in approximately 50% to 60% circumferential narrowing of the abdominal aortic lumen. To prevent infection, penicillin was administered via intramuscular injection within 7 days after surgery.
The rats in the SE group underwent an 8-week exercise training protocol following three days of adaptive swimming training. The SE rats were engaged in swimming for 60 min/day, 5 d/week, for 8 weeks. Swimming was performed with an intensity equivalent to carrying three percent of their body weight, as previously described43. The swimming sessions took place in two large thermostatic swimming buckets with a diameter of 100 cm and a height of 80 cm, maintained at a temperature of 31 ± 1 °C. The water depth was set at 55 cm in separate lanes to prevent the rats from supporting themselves by touching the bottom with their tails. To eliminate the interference of other factors, rats in the CN, sham, and SC groups were placed in water twice a week for 10 min each time.
Echocardiography and heart measurement
After 8 weeks of swimming exercise, 3 rats from each group were selected and underwent heart function evaluation under anaesthetized conditions using appropriate amount of isoflurane. M-mode images were captured to assess various parameters. Left ventricular end-diastolic septum thickness (LVSd), left ventricular end-systolic septum thickness (LVSs), left ventricular end-diastolic diameter (LVIDd), left ventricular end-systolic diameter (LVIDs), left ventricular end-diastolic posterior wall thickness (LVPWd), left ventricular end-systolic posterior wall thickness (LVPWs), left ventricular shortening fraction (FS) and left ventricular ejection fraction (EF) were obtained. Measurements were taken for body weight (BW, mg), heart weight (HW, mg) and left ventricle weight (LW, mg). The HW/BW and LW/BW ratios were calculated as indices to evaluate LVH.
Haematoxylin–eosin (H&E) and masson staining
The rats were humanely sacrificed with an overdose of sodium pentobarbital anesthesia. After the rats were sacrificed, the heart tissues were removed under anaesthesia and fixed in 4% paraformaldehyde for 24 h. Subsequently, the tissues were dehydrated, embedded in paraffin, and sliced into 5-μm thick sections. The sections were stained with H&E and Masson’s trichrome. Images of the heart structures and fibrosis were visualized under a light microscope (OLYMPUS IX73P1F, Japan) and quantified using ImageJ software. To compare myocyte cross-sectional area, 100 cells were measured from 3 heart samples per group.
TUNEL staining
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining was conducted to detect cardiomyocyte. Then, 5-μm thick heart sections and a TUNEL cell apoptosis detection kit (Beyotime Biotechnology, China) were used, following the manufacturer’s instructions. The apoptotic cells with positive TUNEL staining showed a brown colour under an inverted fluorescence microscope (OLYMPUS IX73P1F, Japan).
Transmission electron microscopy (TEM) assay
The left ventricular tissue was cut into approximately 1 mm cubes and fixed with precooled 2.5% glutaraldehyde overnight at 4 °C. Subsequently, the heart tissue was rinsed three times with phosphoric acid buffer (precooled) at room temperature for 20 min each time. Afterwards, the cells were fixed with 1% silver acid (precooled) for 2 h. The tissues were sliced into 60–80 nm sections with an ultramicrotome. Following double staining with uranyl acetate and lead citrate, the samples were dried at 37 °C for 48 h and subsequently observed and photographed using a transmission electron microscope (OLYMPUS FV3000, Japan).
Bioinformatics
RNA was extracted and purified from the left ventricle, and quality control filtering was performed to obtain clean data. RNA whole transcriptome sequencing (RNA-Seq) was conducted by Beijing Genomics Institute (Beijing, China). The analysis, mapping, and mining of the data were conducted using the Dr. Tom multiomics data mining system (https://biosys.bgi.com).
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was employed to identify differential gene enrichment pathways. The differential gene set on the enrichment pathway was further analysed using protein–protein interaction (PPI) relationship association to identify all genes interacting with the differential gene set, resulting in the formation of a second differential gene set. Key driver genes (KDA) within this gene set were screened using key driver analysis, resulting in the identification of ten key driver genes comprising the third gene set. These key genes were compared with the original gene set to facilitate their selection.
Cell line and plasmids
H9C2 cells were purchased from the Cell Bank of the Chinese Academy of Sciences. Adenoviruses overexpressing ndufb10 and control GFP were obtained from Vigene Biosciences (Jinan, China).
The overexpression line used in this study was synthesized by Vigene Biosciences (Jinan, China), and the sequences are shown in the supplementary material.
The control green fluorescent protein (GFP) used in this study is shown in the supplementary Fig. 1.
The sh-ndufb10 used in this study was synthesized by Vigene Biosciences (Jinan, China), and the sequences are shown in the supplementary material.
The sh-control used in this study was synthesized by Vigene Biosciences (Jinan, China), and the sequences are shown in the supplementary material.
Cell culture and adenoviral infection
The frozen cells were retrieved from liquid nitrogen and quickly thawed in a 37 °C water bath. The cell suspension was then transferred to a cell culture flask and cultured in a cell incubator (Shanghai Yiheng Scientific Instrument Co., Ltd.) at 37 °C with 5% CO2. The cells were passaged when they reached approximately 80% confluency and cultured in complete medium consisting of 10% fetal bovine serum (FBS, Gibco, America), 89% Dulbecco’s modified eagle medium (DMEM, Gibco, America), and a 1% streptomycin mixture (Servicebio, China).
The H9C2 cells were randomly divided into six groups: the Ang II 0 h group (CON), Ang II 48 h group (Ang II), Ang II 48 h + sh-control group (sh-GFP + Ang II), Ang II 48 h + sh-ndufb10 group (sh-ndufb10 + Ang II), Ang II 48 h + overexpressedndufb10 control group (Ad-GFP + Ang II) and Ang II 48 h + over-expressedndufb10group (Ad-ndufb10 + Ang II). For Ang II treatment, cells were treated with AngII (100 nmol/L) and 8% FBS DMEM for 48 h, and the CON group was cultured in 8% FBS DMEM for 48 h as a negative control. All H9C2 cells were starved for 24 h before adding Ang II.
A total of 1 × 106 cells were plated in a well of 6-well plates for adenovirus infection. Subsequently, the sh-ndufb10 and Ad-ndufb10 groups were infected with 3 × 108 multiplicity of infection (MOI) adenovirus and incubated for 2 h. Then, 2 ml DMEM with 2% FBS was added to each well, and the culture medium was replaced after 10 h. GFP fluorescence and qPCR were used to monitor the efficiency of infection after 24 h of infection in the sh-ndufb10 group and 48 h of infection in the Ad-ndufb10 group (the control group was treated under the same conditions), and cells with an infection efficiency of > 70% were selected for further experiments.
Mitochondrial analysis
The mitochondrial membrane potential was determined using a JC-1 kit (Beyotime Biotechnology, China) following the manufacturer’s instructions. After staining with JC-1, the ratio of red‒green fluorescence was observed using a confocal laser scanning microscope (OLYMPUS FV3000, Japan) to compare the level of mitochondrial membrane potential. The mitochondrial ROS production was measured using a DCFH-DA fluorescent probe (Solarbio, China). A NAD+/NADH assay kit (Beyotime Biotechnology, China) was used to determine the activity of mitochondrial complex I. The mPTP assay kit (Beyotime Biotechnology, China) and TUNEL assay kit (Beyotime Biotechnology, China) were employed to detect mitochondrial permeability transition pore opening and mitochondrial apoptosis, respectively.
Quantitative reverse transcription-polymerase chain reaction (qRT‒PCR)
Total RNA was extracted from tissues or cells using TRIzol, and cDNA synthesis was performed using the PrimeScript™ RT Master Mix kit (Takara, Osaka, Japan) according to standard protocols. The relative gene expression was measured by real-time PCR using the Applied Biosystems QuantStudioTM5 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). Real-time PCR was conducted according to the provided protocol of the SYBR Premix EX Taq II (Tli RNase H Plus) Kit (Takara, Osaka, Japan). The PCR sequences of the primers used in this study are summarized in Table 3.
Western blotting analysis
Proteins were extracted from frozen heart tissues or cells using radio immunoprecipitation assay (RIPA) buffer supplemented with proteinase and phosphatase inhibitors (Beyotime Biotechnology, China). The protein concentrations in the cleared lysates were measured using bicinchoninic acid (BCA; Beyotime Biotechnology, China) after centrifugation at 12,000×g at 4 °C for 10 min. Automated western immunoblotting was performed using a capillary-based electrophoresis system (Protein Simple, America) following the manufacturer’s instructions. Wes Separation Capillary Cartridges (Protein Simple, America) for 2–40 kDa, 66–440 kDa and 12–230 kDa were used based on the molecular weight of the target proteins44,45,46. A total of 3 μL of protein was loaded onto each capillary, and specific protein levels were identified using specific primary (summarized in Table 4) and secondary antibodies (Protein Simple, America). Chemiluminescence signals were visualized and cropped using Protein Simple Compass software (Protein Simple, America).
Statistics
Each data point is represented as the mean ± SD. All data were analysed using SPSS Statistics 25.0 and GraphPad Prism 9.0 software. One-way ANOVA and Tukey’s post hoc test were performed to compare any two groups, and statistical significance was set at p < 0.05.
Data availability
All data needed to evaluate the conclusions of the paper are present in the paper and/or the Supplementary Materials. The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0005695. Additional data related to this paper are available from the corresponding author on reasonable request.
References
Nakamura, M. & Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407 (2018).
Chan, D. C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287 (2012).
Ferreira, J. C. B. et al. A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats. Nat. Commun. 10, 329 (2019).
Skulachev, V. P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 26, 23–29 (2001).
Bers, D. M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127 (2014).
Tham, Y. K., Bernardo, B. C., Ooi, J. Y. Y., Weeks, K. L. & McMullen, J. R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch Toxicol 89, 1401–1438 (2015).
Brown, D. A. et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 14, 238–250 (2017).
Elorza, A. A. & Soffia, J. P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An integrative view of OXPHOS and mitochondrial life cycle in cardiac mitochondrial physiology. Front. Cell Dev. Biol. 9, 625020 (2021).
Mariño, G., Niso-Santano, M., Baehrecke, E. H. & Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94 (2014).
Abate, M. et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 98, 139–153 (2020).
Scheffer, D. L., Garcia, A. A., Lee, L., Mochly-Rosen, D. & Ferreira, J. C. B. Mitochondrial fusion, fission, and mitophagy in cardiac diseases: Challenges and therapeutic opportunities. Antioxid. Redox Signal. 36, 844–863 (2022).
Friederich, M. W. et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddw431 (2016).
Mellick, G. D., Silburn, P. A., Prince, J. A. & Brookes, A. J. A novel screen for nuclear mitochondrial gene associations with Parkinson’s disease. J. Neural Transm. (Vienna) 111, 191–199 (2004).
Dunham-Snary, K. J. et al. Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 124, 1727–1746 (2019).
Tanoorsaz, S., Behpoor, N. & Tadibi, V. Changes in cardiac levels of caspase-8, Bcl-2 and NT-proBNP following 4 weeks of aerobic exercise in diabetic rats. Int. J. Basic Sci. Med. 2, 172–177 (2017).
Li, H. et al. Exercise training enhances myocardial mitophagy and improves cardiac function via Irisin/FNDC5-PINK1/Parkin pathway in MI mice. Biomedicines 9, 701 (2021).
Zhao, Y., Zhu, Q., Song, W. & Gao, B. Exercise training and dietary restriction affect PINK1/Parkin and Bnip3/Nix-mediated cardiac mitophagy in mice. gpb 37, 657–666 (2018).
Bo, H. et al. Regulation of mitochondrial uncoupling respiration during exercise in rat heart: Role of reactive oxygen species (ROS) and uncoupling protein 2. Free Radic. Biol. Med. 44, 1373–1381 (2008).
Nijholt, K. T., Sánchez-Aguilera, P. I., Voorrips, S. N., de Boer, R. A. & Westenbrink, B. D. Exercise: A molecular tool to boost muscle growth and mitochondrial performance in heart failure?. Eur. J Heart Fail. 24, 287–298 (2022).
Wang, X. et al. Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy. Nat. Commun. 13, 3947 (2022).
Ma, Z., Qi, J., Gao, L. & Zhang, J. Role of exercise on alleviating pressure overload-induced left ventricular dysfunction and remodeling via AMPK-dependent autophagy activation. Int. Heart J. 61, 1022–1033 (2020).
Fernandes, T., Soci, U. P. R. & Oliveira, E. M. Eccentric and concentric cardiac hypertrophy induced by exercise training: MicroRNAs and molecular determinants. Braz. J. Med. Biol. Res. 44, 836–847 (2011).
Liao, J., Li, Y., Zeng, F. & Wu, Y. Regulation of mTOR pathway in exercise-induced cardiac hypertrophy. Int. J. Sports Med. 36, 343–350 (2015).
Meng, X.-X., Yao, M., Zhang, X. D., Xu, H.-X. & Dong, Q. ER stress-induced autophagy in melanoma. Clin. Exp. Pharmacol. Physiol. 42, 811–816 (2015).
Campos, J. C. et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 13, 1304–1317 (2017).
Roh, J., Rhee, J., Chaudhari, V. & Rosenzweig, A. The role of exercise in cardiac aging: From physiology to molecular mechanisms. Circ. Res. 118, 279–295 (2016).
Zhang, Y. et al. MicroRNA-34c-5p provokes isoprenaline-induced cardiac hypertrophy by modulating autophagy via targeting ATG4B. Acta Pharm. Sin. B 12, 2374–2390 (2022).
Sciarretta, S., Maejima, Y., Zablocki, D. & Sadoshima, J. The Role of autophagy in the heart. Annu. Rev. Physiol. 80, 1–26 (2018).
Kubli, D. A. et al. PINK1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS ONE 10, e0130707 (2015).
Zaib, S. et al. Role of mitochondrial membrane potential and lactate dehydrogenase A in apoptosis. ACAMC 22, 2048–2062 (2022).
Lee, G. J., Yan, L., Vatner, D. E. & Vatner, S. F. Mst1 inhibition rescues b1-adrenergic cardiomyopathy by reducing myocyte necrosis and non-myocyte apoptosis rather than myocyte apoptosis. Basic Res. Cardiol. 110, 7 (2015).
French, C. J., Spees, J. L., Taatjes, D. J. & Sobel, B. E. The magnitude and temporal dependence of apoptosis early after myocardial ischemia with or without reperfusion. FASEB J. 23, 1177–1185 (2009).
Shi, X. et al. ndufa7 plays a critical role in cardiac hypertrophy. J. Cell. Mol. Med. 24, 13151–13162 (2020).
Rabinovich-Nikitin, I., Dhingra, R. & Kirshenbaum, L. A. Activation of mitophagy in high-fat diet-induced diabetic cardiomyopathy. Circ. Res. 124, 1288–1290 (2019).
Poznyak, A. V., Ivanova, E. A., Sobenin, I. A., Yet, S.-F. & Orekhov, A. N. The role of mitochondria in cardiovascular diseases. Biology 9, 137 (2020).
Ramachandra, C. J. A., Hernandez-Resendiz, S., Crespo-Avilan, G. E., Lin, Y.-H. & Hausenloy, D. J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 57, 102884 (2020).
Onishi, M., Yamano, K., Sato, M., Matsuda, N. & Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40, e104705 (2021).
Knapp-Wilson, A. et al. Maintenance of complex I and its supercomplexes by NDUF-11 is essential for mitochondrial structure, function and health. J. Cell Sci. 134, jsc258399 (2021).
Onukwufor, J. O. et al. A reversible mitochondrial complex I thiol switch mediates hypoxic avoidance behavior in C. elegans. Nat. Commun. 13, 2403 (2022).
Campos, J. C. et al. Exercise training restores cardiac protein quality control in heart failure. PLoS ONE 7, e52764 (2012).
Emter, C. A. & Baines, C. P. Low-intensity aerobic interval training attenuates pathological left ventricular remodeling and mitochondrial dysfunction in aortic-banded miniature swine. Am. J. Physiol.-Heart Circ. Physiol. 299, H1348–H1356 (2010).
Campos, J. C., Gomes, K. M. S. & Ferreira, J. C. B. Impact of exercise training on redox signaling in cardiovascular diseases. Food Chem. Toxicol. 62, 107–119 (2013).
Ma, Z., Qi, J., Meng, S., Wen, B. & Zhang, J. Swimming exercise training-induced left ventricular hypertrophy involves microRNAs and synergistic regulation of the PI3K/AKT/mTOR signaling pathway. Eur. J. Appl. Physiol. 113, 2473–2486 (2013).
Mahadevan, K. K. et al. KRASG12D inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8+ T cells. Cancer Cell 41, 1606-1620.e8 (2023).
Wang, D. et al. Soluble CD146, a cerebrospinal fluid marker for neuroinflammation, promotes blood-brain barrier dysfunction. Theranostics 10, 231–246 (2020).
Wang, P. et al. Lin28a maintains a subset of adult muscle stem cells in an embryonic-like state. Cell Res. 33, 712–726 (2023).
Acknowledgements
We thank all of the research staff and students who assisted with animal sampling. We thank Xuefang An and Fan Zhang from the Experimental Animal center, Wuhan Institute of Virology for providing technical support.
Funding
This study was supported by the Construction of Virtual Simulation Laboratory for Sports Rehabilitation (Grant no.220502690245032).
Author information
Authors and Affiliations
Contributions
Z. M. designed the project and Y. C. contributed to data analysis and wrote the paper. L. J. developed the experimental design and contributed to data generation. W. X., C. M. and Y. H. conducted the mRNA analysis from cardiac tissue. J.S., R. B. and C. L. analyzed the protein expression in cardiac tissue and contributed to paper writing. Z. M. and Y. C. contributed to the work equally. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All animals were obtained from the Experimental Animal Center of the Three Gorges University (Certificate No. 42010200007182). All animal experimental protocols were approved by the Animal Care and Use Committees of Wuhan Sports University (protocol number 0087–202105-0101).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Z., Cen, Y., Xun, W. et al. Exercise enhances cardiomyocyte mitochondrial homeostasis to alleviate left ventricular dysfunction in pressure overload induced remodelling. Sci Rep 15, 11698 (2025). https://doi.org/10.1038/s41598-025-95637-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-95637-z
