
Abstract
Immunotherapy has emerged as a new standard of care for certain cancer patients with specific cellular and molecular makeups. However, there is still an unmet need for ex vivo models able to readily assess the effectiveness of immunotherapeutic treatments in a high-throughput and patient-specific manner. To address this issue, we have developed a microarrayed system of patient-derived tumoroids with recreated immune microenvironments that are optimized for the high-content evaluation of tumor-infiltrating lymphocyte functionality. Here we show that this system offers unprecedented opportunities to evaluate tumor immunogenicity, characterize the response to immunomodulators, and explore novel approaches for personalized immuno-oncology.
Similar content being viewed by others

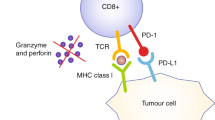

After the initial success in the treatment of melanoma, immunotherapy has quickly arisen as an effective treatment for several types of solid cancers, including colorectal cancer (CRC)1. In the latter, however, response rates remain limited to a fraction of the cases for reasons not yet fully understood2,3,4. This underscores the need for in vitro experimental systems capable of efficient patient-specific screenings for both therapy selection and response prediction. Over the last decade, diverse three-dimensional (3D) cell culture systems have been developed to that end5,6,7. Among them, tumor organoids (tumoroids) have arguably become the most widely adopted model as they can faithfully mimic key aspects of tumor biology, including histopathological features, cellular heterogeneity, gene expression patterns, and responses to specific chemotherapies8,9,10. Despite these advantages, conventional tumoroid cultures are low-throughput, topologically heterogeneous, and require high quantities of ill-defined extracellular matrices such as Matrigel. For these reasons, here we sought to develop a 3D model able to mitigate these shortcomings and escalate the potential for ex vivo assessment of tumor-infiltrating lymphocyte (TIL) functionality.
We based our tumoroid culture platform on hydrogel films comprising arrays of 500-μm round-bottom microcavities located at the bottom of 96-well plate wells (Fig. 1a). Unlike conventional organoid cultures, this system allows for highly efficient cell aggregation at pre-defined positions in a matrix-free setting, leading to exceptionally homogeneous organoid formation (Fig. 1a, b). This arrangement allows for enhanced cell–cell interactions among the cells inside a microcavity, reliable tumoroid tracking over time, and high-throughput and high-resolution imaging analyses (Fig. 1a, b). To validate the suitability of this platform for the assessment of T cell anti-tumor activity, we used the pmel-1 mouse model. These animals bear T cells with a transgenic TCR receptor able to recognize the gp100 antigen expressed by murine B16 melanomas, ensuring immune cell responsiveness and tumor cell killing. Thus, we seeded B16-F10 melanoma cells, allowed tumoroid formation, and then added pmel-1 T cells at different effector-to-target ratios (Fig. 1c). We confirmed that the microcavity system readily allowed the high-resolution monitorization of T cell migration and reactivity, together with tumoroid size and viability, in multiple homogeneous replicates generated with minimal effort (Fig. 1d, e). To quantify the killing efficacy of T cells, we stained dead cells with propidium iodide (PI) and found a direct correlation between the cytotoxic activity and the effector-to-target cell ratios, demonstrating the sensitivity and specificity of the system (Fig. 1f).

a Illustration of the differences between standard (top) and microarrayed (bottom) culture systems for tumoroids. b Representative brightfield images of standard (left) and microarrayed (right) tumoroid cultures. Scale bar, 500 µm. c Scheme of the experimental workflow followed in this study. d Representative images of microarrayed co-cultures of tumoroids and T cells at the indicated ratios (left) and time (top). Scale bar, 250 µm. e Tumoroid area over time at the indicated cell ratios and time. **P < 0.01, ***P < 0.001 (Kruskal–Wallis and Dunn’s tests, n = 38 (top), and ANOVA and Tukey’s tests, n = 37 (bottom)). f Measurement of the PI signal at day 2 in the indicated cell ratio conditions. **P < 0.01, ***P < 0.001 (Kruskal–Wallis and Dunn’s tests, n = 32). In e, f, data represent mean ± SEM.
To evaluate the translational potential of this platform, we then focused on the assessment of T-cell activity in a cohort of five patient-derived colorectal cancer samples with diverse clinical and mutational profiles (Supplementary Table 1, Supplementary Fig. 1a). After isolation and expansion, the TILs obtained from these samples were constituted by over 85% T cells, mostly CD8+ with sparse presence of CD4+ and NKT cells (Supplementary Fig. 1b), expressing different degrees of activation markers (CD25, CD69, HLA-DR) (Supplementary Fig. 1c) and inhibitory immunoreceptors (CTLA-4, PD-1) (Supplementary Fig. 1d). They also displayed diverse transcriptional reactions upon co-culture with their autologous tumoroid and microenvironmental cells (Supplementary Fig. 1e). To make sure that they were susceptible to antigen-mediated stimulation, we induced TCR signaling with α-CD3 and α-CD28 antibodies as a positive control. Under these conditions, we observed that all lines were capable of eliciting a cytotoxic response (Fig. 2a, b). To evaluate autonomous antigen recognition and tumor targeting, we next used conditions deprived of α-CD3 and α-CD28, where we found that only the TILs from 2 patients (#MS and #NW) were able to kill their autologous tumoroids (Fig. 2a, b). This response correlated with an increased capability of the TILs to migrate towards the tumoroid (Supplementary Figs. 2, 3a). To validate the specificity of these results, we labeled the tumoroids with a luciferase reporter and cultured them with autologous TILs in Matrigel domes on standard 96-well plates. Notably, compared to the microcavity platform, these conventional tumoroid cultures produced much more heterogeneous systems with poor cell aggregation and low imaging resolution (Supplementary Fig. 3b–d). Nevertheless, based on CRC luminescence we could confirm that the TILs from patients #MS and #NW inhibited tumor cell growth in an autonomous manner (Supplementary Fig. 3c, d). As expected, this anti-tumor activity was enhanced upon α-CD3/CD28 co-stimulation (Supplementary Fig. 3c, d). Conversely, we did not detect CRC growth impairment by the TILs in patient #GQ (Supplementary Fig. 3b). Taken together, these results indicate that the microcavity system can reproduce the results of standard culture models with higher resolution and throughput.

a Representative images of microarrayed patient-derived co-cultures of CRC tumoroids and autologous TILs in the indicated conditions and patients after 3 days of culture. Unless otherwise indicated, 10:1 (TIL:CRC) cell ratios are shown. Culture conditions: autonomous activation (IL-2), ectopic activation (IL-2 + α-CD3/CD28), immunotherapy (IL-2 + α-CD3/CD28 + α-PD-1/CTLA-4). Scale bar, 500 µm. b Quantification of PI signal in the indicated patients and conditions from the experiment shown in panel (a). NS, not significant; P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001 (ANOVA and Tukey’s tests, n = 62, 61, 68, 59, and 53 for patients #TF, #GQ, #MS, #NW, and #NS, respectively). c Scheme depicting the recreation of complex tumor microenvironments inside microcavities. DC dendritic cells, TIL tumor-infiltrating lymphocytes, CRC colorectal cancer, CAF cancer-associated fibroblasts. d Representative images of microarrayed CRC tumoroids integrated in complex tumor microenvironments. Scale bar, 250 µm. e Luminescence-based measurement of the promotion of TIL killing activity induced by CD40-mediated DC activation in CRC tumoroids derived from patient #TF. ***P < 0.001 (ANOVA and Sidak’s tests, n = 6). In b and e, data represent mean ± SEM.
To evaluate the responses to immunotherapeutic agents currently used in the clinic, we next treated the microarrayed tumoroids with the immune checkpoint blockade (ICB) antibodies Nivolumab (PD-1 inhibitor) and Ipilimumab (CTLA-4 inhibitor). In keeping with the clinical evidence for CRC11, this translated into increased cytotoxicity only in a small fraction of the patients (1 out 5, patient #NW) (Fig. 2b; Supplementary Figs. 2c, d; 3a). To formally prove that this increased cytotoxicity was associated with enhanced T cell activation, we profiled the release of Granzyme B (GrB), Interferon-γ (IFNγ), and tumor necrosis factor (TNF) in the microcavities. As expected, we found that both higher effector-to-target T cell ratios and co-stimulation with α-CD3/CD28 promoted cytokine production (Supplementary Fig. 3e). This response was enhanced by immune checkpoint blockade agents only in the case of TILs derived from patient #NW, in line with our previous results (Supplementary Fig. 3e). We also observed discrepancies between cytokine production (Supplementary Fig. 3e) and killing activity (Fig. 2b) in some patients, suggesting that TIL cytotoxicity was not solely due to the production of these cytokines, but additional patient-specific mechanisms.
Since immunotherapy involves additional cellular players besides TILs and the microcavities are particularly effective in promoting cell aggregation, we developed tumoroids with complex microenvironments to explore alternative ways to engage TILs in patients not responsive to standard ICB therapy. Specifically, we co-cultured CRC cells with TILs, cancer-associated fibroblasts, and monocyte-derived dendritic cells (Fig. 2c, Supplementary Fig. 4a, b). These different cell types formed highly integrated heterotypic micro-tissues inside the microcavities, where their internal composition could be readily tracked (Fig. 2d). Given that CD40 activation in dendritic cells is emerging as a promising approach to promote TIL functionality in immunologically cold tumors12, we treated these tumoroids with CD40 ligand. Supporting the current evidence12, this approach was able to enhance TIL activity against tumor cells even in patients not responding to PD-1 blockade (Fig. 2e, Supplementary Fig. 4c, d). This occurred in a DC-dependent manner (Supplementary Fig. 4e), both of primary and cell line origin (Supplementary Fig. 4f). Collectively, these results highlight the versatility of the microcavity-based model for the assessment of T cell responsiveness in different immunomodulatory contexts.
Altogether, in this work, we provide a unique experimental setup in which a large number of uniform tumoroids can be robustly examined at the single-tumoroid level. Unlike other co-culture systems that require the use of complex extracellular matrices or do not allow precise control over tumoroid distribution and size, our platform provides unparalleled resolution of tumoroid-TIL interactions. Building on these features, we show proof-of-concept data for the application of this system for immuno-oncology studies. Importantly, we acknowledge that expanding our study to additional immunotherapy-responsive patient cohorts will be required to fully assess the translational correlation and prospects of the system. In addition, (i) the collection of biopsies from multiple tumor sites to account for tumor heterogeneity, (ii) the optimization of culture conditions to prevent cell drift in vitro, (iii) the adjustment of TIL:CRC cell ratios according to the native tumor-specific proportions, and (iv) the testing of additional immunomodulatory conditions (such as immunotherapy without α-CD3/CD28 stimulation) will be required to provide deeper biological insights. Nevertheless, our results already demonstrate that the microcavity platform allows for the stratification of colorectal cancer patients according to both tumor-infiltrating lymphocyte reactivity and response to immune checkpoint blockade therapy. Significantly, our findings are aligned with the current clinical evidence13, since we detected the strongest TIL activity in patients that have either microsatellite instability (#MS) or pathogenic mutations in DNA mismatch repair genes (#NW, mutation in MSH6) (Supplementary Table 1, Supplementary Fig. 1a). We also show that this platform can help illuminate heterotypic interactions in the tumor microenvironment that are exploitable for immunotherapy, further supporting its suitability as a screening tool for personalized immunotherapy approaches. Once identified, these can be further characterized in other culture platforms that, while being lower-throughput, are particularly well suited for long-term experiments, such as the recently developed CRC mini-colons14,15.
Methods
Ethics statement
The use of patient samples was conducted in accordance with the provisions of the Declaration of Helsinki. It was reviewed and approved by the ethics committee of the Canton of Vaud and the Centre Hospitalier Universitaire Vaudois (CHUV), under the protocol CHUV_DO_CTE_TRP_0001_2017. All patient samples have been collected with a priori patient consent.
Cell source and isolation
B16-F10 melanoma cells and pre-activated Pmel-1 TCR transgenic T-cells were received as a kind gift from the Laboratory of Biomaterials for Immunoengineering (EPFL). Colorectal cancer cells, cancer-associated fibroblasts, tumor-infiltrating lymphocytes, and peripheral monocytes were isolated from matched CRC biopsies and peripheral blood obtained from the Centre Hospitalier Universitaire Vaudois (CHUV) as indicated here (Broguiere et al.16). Briefly, the CRC biopsy was mechanically dissociated with a scalpel, digested in 0.1% (W/V) collagenase I (Thermo Fisher Scientific, Catalog No. 17100-017) for 30 min at 37 °C, and filtered through a 70-μm strainer. The remaining undigested pieces were further incubated with TrypLE Express (Thermo Fisher Scientific, Catalog No. 12605028) for 10 min at 37 °C and processed as indicated before. The resulting cells were plated in three different conditions: (i) Matrigel (Corning, Catalog No. 356231) embedding in a panel of CRC media17 (CRC fraction), (ii) type I collagen (REPROCELL, Catalog No. KKN-IAC-50)-coated flasks with CAF medium (see below) (CAF fraction), and (iii) round bottom ultra-low attachment 96-well plates (Corning, Catalog No. 7007) with TIL medium (see below) supplemented for 72 h with α-CD3 (1 μg ml–1, BioLegend, Catalog No. 317326) and α-CD28 (1 μg ml–1, BioLegend, Catalog No. 302934) antibodies (TIL fraction). This media selection strategy led to pure CRC, CAF, and TIL populations in 2–4 weeks (Broguiere et al.16, https://doi.org/10.1101/2023.11.01.565169). Overall, a ~80% success rate (25/31) was obtained for CRC + TME derivation from CRC biopsies (Broguiere et al.16, https://doi.org/10.1101/2023.11.01.565169). Monocytes were isolated using Lymphoprep (STEMCELL Technologies, Catalog No. 07851) for peripheral blood mononuclear cell (PBMC) purification, followed by CD14-based magnetic-activated cell sorting (Miltenyi Biotec, Catalog No. 130-050-201), according to the manufacturer’s instructions. Five patient-derived lines representative of different genomic profiles were selected for this study (Supplementary Table 1, Supplementary Fig. 1a). THP-1 monocytes were purchased from the ATCC (ATCC, Catalog No. TIB-202).
Molecular characterizations
All patient-derived cells used in this study have been deeply characterized in the context of a large single-cell RNA-Seq atlasing effort (https://doi.org/10.1101/2023.11.01.565169). All single-cell RNA-Seq data have been obtained from the mentioned study. All the plots based on single-cell data have been generated using the CRC-TME Atlas portal (https://crc-tme.com/), which contains a set of interactive tools for the exploration of the gene expression data from the mentioned study. For exome sequencing, genomic DNA was extracted with the PureLink™ Genomic DNA Mini Kit (Thermo Fisher Scientific, Catalog No. K182002) according to the manufacturer’s instructions. Exome sequencing was carried out by BGI Genomics (Hong Kong) at 100× depth using 150 bp paired-end reads. These reads were mapped to the human genome GRCh38 using BWA-MEM (v0.7.17). Samtools (v1.9) and the Ensembl Variant Effect Predictor (v100.2) were used for variant calling and annotation, respectively. Microsatellite instability was assessed at the Centre Hospitalier Universitaire Vaudois (CHUV) using the VENTANA MMR IHC Panel (Roche Diagnostics). In case of uncertainty, the samples were analyzed by pyrosequencing for microsatellites and by next-generation sequencing with an in-house developed gene panel.
Cell culture
B16-F10 melanoma cells were cultured in DMEM (Thermo Fisher Scientific, Catalog No. 61965059) supplemented with 10% FBS (Thermo Fisher Scientific, Catalog No. 10500064). CRC organoids were embedded in growth factor reduced Matrigel (Corning, Catalog No. 356231) and cultured in Advanced DMEM/F-12 (Thermo Fisher Scientific, Catalog No. 12634028) supplemented with 1× GlutaMAX (Thermo Fisher Scientific, Catalog No. 35050038), 10 mM HEPES (Thermo Fisher Scientific, Catalog No. 15630056), 100 μg ml−1 penicillin–streptomycin (Thermo Fisher Scientific, Catalog No. 15140122), 1 × B-27 supplement (Thermo Fisher Scientific, Catalog No. 17504001), 1 mM N-acetylcysteine (Sigma-Aldrich, Catalog No. A9165), and 50 μg ml−1 Primocin (InvivoGen, Catalog No. ant-pm-2). Cancer-associated fibroblasts were grown in EGM-2 MV microvascular endothelial cell growth medium-2 (Lonza, Catalog No. CC-3202). Tumor-infiltrating lymphocytes were cultured in RPMI-1640 (Thermo Fisher Scientific, Catalog No. 61870036) supplemented with 1× GlutaMAX, 10 mM HEPES, 100 μg ml−1 penicillin–streptomycin, 50 μM 2-mercaptoethanol (Thermo Fisher Scientific, Catalog No. 31350010), 10% FBS, and 1 μg ml−1 recombinant human IL-2 (PeproTech, Catalog No. 200-02). The same medium was used to culture monocytes but in the absence of IL-2. For dendritic cell differentiation, monocytes were cultured in the presence of 250 ng ml−1 IL-4 (PeproTech, Catalog No. 200-04) and 250 ng ml−1 GM-CSF (PeproTech, Catalog No. 300-03) for 5 days. Unless otherwise indicated, co-cultures were maintained in a 1:1 proportion of CRC and TIL media. Cells were tested for mycoplasma before cryopreservation and in randomized routine checks using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza, Catalog No. LT07-705).
Tumor cell aggregation and co-culture establishment
Tumor cells were dissociated using TrypLE Express, filtered (40 μm), and stained with CellTrace™ Far Red (Thermo Fisher Scientific, Catalog No. C34572) according to the manufacturer’s instructions. Microcavity arrays were manufactured as indicated before18 (SUN Biosciences, Catalog No. Gri3D-96P-S-24-500) and equilibrated by adding 200 μl of the appropriate medium (see above) in each well (comprising 73 microcavities) for a 30-min incubation at 37 °C. After removing the equilibration medium, around 3 × 104 cells per well (~400 cells per microcavity) were resuspended in 50 μl of medium and directly seeded onto the microcavities, allowing the cells to sediment for 30 min. After this time, 150 μl of the appropriate medium (see above) was added to the side chamber of each well. Tumoroids were formed and incubated for the desired number of days before co-culture with additional cells. T cells were collected and stained with CFSE (Cayman Chemical, Catalog No. 10009853) according to the manufacturer’s instructions. They were then added to the microcavity system, as indicated above, at the indicated effector-to-target ratios based on the initial cancer cell numbers. T cell activation was achieved using α-CD3 (1 μg ml–1, BioLegend, Catalog No. 317326) and α-CD28 (1 μg ml–1, BioLegend, Catalog No. 302934) antibodies. For ICB therapy, the PD-1 and CTLA-4 inhibitors were added at a concentration of 20 and 10 μg ml–1, respectively. To measure tumor killing by TILs, propidium iodide (Sigma-Aldrich, Catalog No. 81845) was added to each well at 1 μg ml–1. For complex tumoroid formation, cancer-associated fibroblasts and dendritic cells were stained with CellTracker™ Orange (Thermo Fisher Scientific, Catalog No. C2927) and Cytopainter (Abcam, Catalog No. ab176726), respectively, before inclusion in the microwells as indicated for the other cell types. For these complex tumoroids, all cells were seeded at the same time with a ratio of 1:2:0.25:1 for CRC:TIL:CAF:DC. Dendritic cell activation was induced using soluble CD40 ligand at 100 ng ml–1 (PeproTech, Catalog No. 310-02) once the co-culture with the rest of the cell types had been established.
Microscopy and image analysis
Brightfield and fluorescent imaging were performed using a Nikon Eclipse Ti2 inverted microscope system equipped with a ×4/0.13 NA, ×10/0.30 NA, and ×40/0.3 NA air objectives and a DS-Qi2 camera (Nikon Corporation) or an INCell Analyzer 2200 device (GE Healthcare), with a large field-of-view sCMOS camera and a ×4/0.2 NA objective. ImageJ (NIH) was then used to process the images and create maximal intensity projections of the 3D stacks. Brightfield images were used to define each microcavity of interest by applying an intermode threshold, followed by binary processing (dilate, close, fill holes) to represent each microcavity as a black circle. The analyze particles function was then used to detect every circle and store them as region of interest (ROI). When the automatic detection was not possible, black circles (radius of 250 µm) were added manually on the microcavity of interest, before processing with the same thresholding and to analyze particle function. Microcavities containing no cyst, more than one, or large debris were not included in the analysis. Tumoroid area was measured using a threshold on the far-red channel and the analyze particles function. PI intensity was measured in an area of interest 50 µm larger than the tumoroids. To define the extended perimeter, the tumoroids were thresholded as before, and the binary function dilate was used (60 times, count at 3). The analyzed particle function was then necessary to detect each area and save them as ROI. Before measuring PI intensity, compensation was required due to the spectral overlap with the green fluorescence (CFSE). First, a background subtraction (rolling ball radius of 50 px) was performed on both fluorescent channels, then the image calculator function was used to subtract the green intensity (CFSE) from the red channel (PI). The resulting compensated image was used to measure the PI mean intensity within each area of interest. The spread of T cells around tumoroids was measured by computing the area of spread around the tumoroids and the radius of T cells around all tumoroids within a well.
Luciferase-based quantification of T cell killing activity
Lentiviral particles carrying a luciferase reporter were produced at the EPFL Gene Therapy Facility using the pHIV-Luc-ZsGreen plasmid (Addgene, Catalog No. 39196). For transduction, CRC organoids were dissociated into single cells using TrypLE, washed with basal medium 10% FBS, and resuspended in culture medium containing ~1000 ng(p24) ml–1 of lentiviral particles plus 8 μg ml–1 polybrene (Sigma-Aldrich, Catalog No. TR-1003-G). Cells were then centrifuged at 600 × g for 60 min at room temperature and incubated for 6 h at 37 °C. After incubation, they were collected and plated. One week after infection, successfully transduced CRC cells (ZsGreen+) were sorted in a FACSAria Fusion flow cytometer (BD Biosciences). For the monitorization of CRC cell viability, transduced CRC organoids were co-cultured with the desired cells (same conditions as indicated above) in black-wall 96-well plates (Corning, Catalog No. CLS3603-48EA). Live-cell luciferase readings were performed by adding 150 μg ml–1 luciferine (GOLDBIO, Catalog No. LUCK-500) and measuring luminescence in a Tecan Infinite F500 reader (Tecan).
Determination of cytokine production
Secreted interferon-gamma, tumor necrosis factor, and granzyme b were measured in the supernatant of microcavities using Cytometric Bead Array (CBA) assays (BD Biosciences, Catalog Nos. 558264, 560304, 558273, and 560111) according to the manufacturer’s instructions. Samples were acquired in a BD LSR II cell analyzer (BD Biosciences), and results were analyzed using FlowJo software (BD Biosciences).
Determination of monocyte differentiation
Differentiated monocytes were collected by strong pipetting followed by gentle TryPLE digestion (1 min at 37 °C) of the remaining adherent cells, spun down at 400 × g for 4 min, and incubated in PBS with 2% FBS for 30 min on ice with antibodies for CD11c (APC-Cy7) (1:200, BioLegend, Catalog No. 337218) or HLA-DR (PE) (1:50, Miltenyi Biotec, Catalog No. 130-111-942). Cells were then washed three times in PBS with 2% FBS, acquired in an LSRFortessaTM Cell Analyzer (BD Biosciences), and analyzed using FlowJo software (BD Biosciences).
Statistics
Data normality and equality of variances were analyzed with Shapiro–Wilk and Bartlett’s tests, respectively. Parametric distributions were analyzed using the Student’s t-test (when comparing two experimental groups) or ANOVA followed by Dunnett’s test (when comparing more than two experimental groups to a single reference group) or Tukey’s HSD test (when comparing more than two experimental groups with every other group). Sidak’s multiple comparison test was used when comparing different sets of means. Nonparametric distributions were analyzed using the Kruskal–Wallis followed by Dunn’s tests. In all cases, values were considered significant when P ≤ 0.05. Data obtained are given as the mean ± the standard error of the mean (SEM).
Data availability
All patient-derived cells used in this study have been deeply characterized in the context of a large single-cell RNA-Seq atlasing effort (https://doi.org/10.1101/2023.11.01.565169). All single-cell RNA and exome sequencing data have been obtained from the mentioned study and will be available upon publication of the latter. All the plots based on single-cell data have been generated using the CRC-TME Atlas portal (https://crc-tme.com/). All other relevant data are available from the authors.
Code availability
No custom code was used. All the plots based on single-cell data have been generated using the CRC-TME Atlas portal (https://crc-tme.com/).
References
Emens, L. A. et al. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 81, 116–129 (2017).
Wieder, T., Eigentler, T., Brenner, E. & Röcken, M. Immune checkpoint blockade therapy. J. Allergy Clin. Immunol. 142, 1403–1414 (2018).
Jenkins, R. W., Barbie, D. A. & Flaherty, K. T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16 (2018).
Sahin, I. H. et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 121, 809–818 (2019).
Aref, A. R. et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143 (2018).
Neal, J. T. et al. Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–1988.e1916 (2018).
Yuki, K., Cheng, N., Nakano, M. & Kuo, C. J. Organoid models of tumor immunology. Trends Immunol. 41, 652–664 (2020).
van de Wetering, M. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015).
Ooft, S. N. et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aay2574 (2019).
Vlachogiannis, G. et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018).
Sveen, A., Kopetz, S. & Lothe, R. A. Biomarker-guided therapy for colorectal cancer: strength in complexity. Nat. Rev. Clin. Oncol. 17, 11–32 (2020).
Vonderheide, R. H. CD40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med. 71, 47–58 (2020).
Bever, K. M. & Le, D. T. DNA repair defects and implications for immunotherapy. J. Clin. Investig. 128, 4236–4242 (2018).
Lorenzo-Martín, L. F. et al. Spatiotemporally resolved colorectal oncogenesis in mini-colons ex vivo. Nature https://doi.org/10.1038/s41586-024-07330-2 (2024).
Lorenzo-Martín, L. F. et al. Patient-derived mini-colons enable long-term modeling of tumor-microenvironment complexity. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02301-4 (2024).
Broguiere, N. et al. Dissection and reconstruction of the colorectal cancer tumor microenvironment. bioRxiv 2023.11.01.565169; https://doi.org/10.1101/2023.11.01.565169 (2023)
Fujii, M. et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18, 827–838 (2016).
Brandenberg, N. et al. High-throughput automated organoid culture via stem-cell aggregation in microcavity arrays. Nat. Biomed. Eng. 4, 863–874 (2020).
Acknowledgements
We thank L. Tang and K. Lei for providing cells for the mouse assay validations, S. Li for helping with CRC sample handling, F. Kuttler and C. Claus for scientific discussions and inputs on the manuscript, and M. Gueye and I. Marten for helping with cell culture. We acknowledge support from the following EPFL core facilities: BIOP, FCCF, and BSF. This work was funded by the Personalized Health and Related Technologies (PHRT) Initiative from the Swiss Federal Institutes of Technology Board and EPFL.
Author information
Authors and Affiliations
Contributions
D.D. designed experiments, carried out experimental work, analyzed the data, and participated in manuscript writing. L.F.L.-M. designed experiments, carried out experimental work, analyzed the data, and wrote the manuscript. F.R. conceived the initial ideas and performed the founding experiments. N.B. spearheaded the establishment of the CRC and tumor microenvironment cell lines. L.T. helped with experimental work. S.R. (Ragusa) helped with CRC biobank establishment and tumoroid characterization. N.B. and S.H. developed the microcavity platform. S.R. (Rusakiewicz) and D.S. established the T-cell protocols. K.H. provided human CRC samples. G.C. contributed to the experimental design. M.P.L. conceived the work, designed experiments, and carried out the final editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The EPFL has filed for patent protection on the hydrogel microcavity array device and organoid array technologies described here, and M.P.L., N.B., and S.H. are named as inventors on those patent applications. M.P.L., N.B., and S.H. are shareholders in SUN Bioscience SA and DOPPL SA, which are commercializing those patents. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dutta, D., Lorenzo-Martín, L.F., Rivest, F. et al. Probing the killing potency of tumor-infiltrating lymphocytes on microarrayed colorectal cancer tumoroids. npj Precis. Onc. 8, 179 (2024). https://doi.org/10.1038/s41698-024-00661-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00661-3
