
Abstract
Drug-resistant tuberculosis (TB) continues to challenge treatment options, necessitating the exploration of new compounds of novel targets. The mycobacterial respiratory complex cytochrome bc1:aa3 has emerged as a promising target, exemplified by the success of first-in-class inhibitor Q203 in phase 2 clinical trials. However, to fully exploit the potential of this target and to identify the best-in-class inhibitor more compounds need evaluation. Here, we introduce JNJ-2901, a novel Q203 analogue, that demonstrates activity against multidrug-resistant M. tuberculosis clinical strains at sub-nanomolar concentration and 4-log reduction in bacterial burden in a mouse model of TB infection. Inhibitory studies on purified enzymes validate the nanomolar inhibitions observed in mycobacterial cells. Additionally, cryo-EM structure analysis of cytochrome bc1:aa3 bound to JNJ-2901 reveals the binding pocket at the menaquinol oxidation site (Qp), akin to other substate analogue inhibitors like Q203 and TB47. Validation of the binding site is further achieved by generating and isolating the JNJ-2901 resistant mutations in M. tuberculosis, followed by purification and resistance analysis of the resistant cytochrome bc1:aa3 complex. Our comprehensive work lays the foundation for further clinical validations of JNJ-2901.
Similar content being viewed by others

Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a significant threat to global health, with over 10.6 million individuals falling ill and more than 1.3 million deaths recorded in 20221, underscoring its profound impact on global health. The prevalence of drug-resistant (MDR) and extensively drug-resistant (XDR) TB necessitates longer treatment durations (18-20 months) with multiple drugs and often leads to poor treatment outcomes compared to drug-sensitive TB cases. Despite recent MDR/XDR-TB treatment breakthroughs, such as the use of bedaquiline, pretomanid, and linezolid with or without moxifloxacin (BPaL/BPaLM)2, the emergence of bedaquiline-resistant M. tuberculosis strains could pose a threat to successful treatment outcomes3,4,5,6. There is an urgent need for the development of new therapies and strategies to combat the escalating challenge that requires the identification of novel compounds and targets along with a thorough understanding of their modes of action and resistance development.
Bedaquiline is a key component of the new TB treatment regimens currently undergoing development7,8,9,10. It inhibits the F1F0 ATP synthase enzyme of the mycobacterial electron transport chain11,12,13. The discovery of bedaquiline has sparked interest in other enzymes of the electron transport chain as novel targets for TB drug discovery14,15,16. Among these targets, the cytochrome bc1:aa3 enzyme complex (cytochrome bc) stands out, as evidenced by the progress of its inhibitors like Q20317 and TB4718. Q203 has shown to be safe in a phase 1b clinical trial19 and shown good results in the phase 2 clinical trials20, while TB47 is in preclinical stage (https://www.newtbdrugs.org/pipeline/clinical). Cytochrome bc is an intermediate complex in the electron transport chain and functions as a terminal electron acceptor, converting oxygen to water and contributing to the proton motive force, required for ATP generation for bacterial survival21,22,23. Both the molecules Q203 and TB47 are substrate analogs (menaquinone) and function by preventing substrate binding24,25. Another line of evidence supporting cytochrome bc as a promising drug target was found in a genome-wide CRISPRi screen in M. tuberculosis, where it was found to be one of the top vulnerable targets26.
Considering the high attrition rate of the drug discovery process and to increase the chance of bringing cytochrome bc inhibitors to the treatment stage, additional inhibitors must be added to the pipeline, and consequently, multiple efforts are made to search for novel inhibitors of cytochrome bc, see27,28,29,30 and further work reviewed in Wani et al.31 and Bajeli et al.32. Here, we introduce JNJ-2901, a novel analog of Q203 and inhibitor of cytochrome bc. We show that JNJ-2901 inhibits the growth of clinical multidrug-resistant M. tuberculosis strains at sub-nanomolar concentrations and reduces lung colony forming unit (CFU) by 4 logs in a mouse model of M. tuberculosis infection. At the enzymatic level, we confirm JNJ-2901’s nano-molar inhibitory potency. By determining the cryo-EM structure of a modified cytochrome bc from M. smegmatis in which the active site has been made identical to that of M. tuberculosis we show that JNJ-2901 occupies the menaquinol oxidation site (Qp) in a manner analogous to Q203 and TB47. Furthermore, we generated laboratory mutants of JNJ-2901 in M. tuberculosis and identified the resistant mutations within the binding pocket. To gain further insight into the resistant mechanism, we purified the resistant enzyme complex and analyzed the resistance at the enzymatic level. These resistant enzymes will serve as tools to screen novel inhibitors targeting resistant enzyme complexes. Our comprehensive work establishes that JNJ-2901 is an effective, potent inhibitor of M. tuberculosis cytochrome bc with the potential for further development towards a novel antibiotic in the continuing battle against drug-resistant TB.
Results
JNJ-2901 inhibits bacterial growth in-vitro and in-vivo
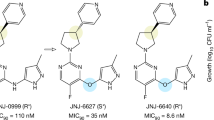
JNJ-2901 is a novel analog of the M. tuberculosis cytochrome bc inhibitor Q20317 (also known as Telacebec) and TB4718 (Fig. 1a). To measure its potential in reducing bacterial growth, we tested JNJ-2901 on a panel of 18 clinical isolates of M. tuberculosis with different antibiotic resistance profiles for commonly used antibiotics (Table 1). For all 18 clinical strains, we found that JNJ-2901 is a potent inhibitor of growth in the Middlebrook 7H9 medium with a MIC of ≤0.5 μg/L (≤1 nM). Interestingly, the H37Rv and Erdman reference strains are less sensitive to JNJ-2901 (MIC of >8 μg/L, >16 nM). The reduced susceptibility of H37Rv for inhibition of cytochrome bc was also observed by others26,33,34. Furthermore, it was shown that in this strain, the deletion or inhibition of the cytochrome bd oxidase results in restoration of cytochrome bc sensitivity33,34,35, suggesting that cytochrome bd could take over the bulk transport of protons when cytochrome bc is inhibited. Also in our hands, we find that a deletion strain for cytochrome bd, M. tuberculosis H37Rv (H37Rv-ΔcydAB), shows restored sensitivity for JNJ-2901, with an MIC50 of 2.5 ± 0.1 nM, similar to Q203 (MIC50 of 2.0 ± 0.1 nM) (Fig. 1b).

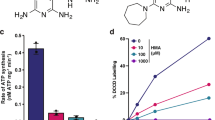
a Chemical structures of JNJ-2901, Q203, and TB47. b Inhibitory profile of Q203 and JNJ-2901 on M. tuberculosis H37Rv-ΔcydAB grown in 7H9 medium. MIC50 values Q203: 2.0 ± 0.1, JNJ-2901: 2.5 ± 0.1. c Inhibitory activity of JNJ-2901 on membranes prepared from M. smegmatis wild-type strain, a cytochrome bc knockout strain (ΔqcrCAB) or a cytochrome bd knockout strain (ΔcydAB). d Survival of Mtb H37Rv cytochrome bd knockout strain (ΔcydAB) in an acute mouse model upon exposure with bedaquiline (BDQ) or JNJ-2901 (mpk = milligram per kilogram). Each group contained 6 mice, and error bars indicate standard deviation. e Survival of Mtb H37Rv-ΔcydAB in a chronic mouse model upon exposure with bedaquiline (BDQ) or JNJ-2901 (mpk = milligram per kilogram).
To further validate that JNJ-2901 indeed targets cytochrome bc, we measured the oxygen consumption rate using a Clark-type electrode on isolated membranes of either the wildtype M. smegmatis strain MC215536, a cytochrome bc knockout strain (M. smegmatis ΔqcrCAB)37 or a cytochrome bd knockout strain (M. smegmatis ΔcydAB)38. Here, we find that JNJ-2901 inhibits oxygen consumption in both the wild-type strain and the cytochrome bd knockout strain with an IC50 of 89 ± 54 and 60 ± 26 nM, respectively (Fig. 1c), comparable to previous values for inhibition by Q203 on isolated membranes from M. smegmatis and M. tuberculosis (IC50 ~ 20 nM)35. In contrast, the cytochrome bc knockout strain (ΔqcrCAB) that depends entirely on cytochrome bd is resistant to JNJ-2901, with a modest 20% reduction of activity at 10 μM. Interestingly, while Q203 and JNJ-2901 show potent inhibition of oxygen consumption on isolated membranes from M. smegmatis, whole cell M. smegmatis is mostly resistant to inhibition by Q203 (MIC > 50 μM)35 while even a cytochrome bd knockout strain of M. smegmatis showed little sensitivity to Q203 (MIC ~ 2.5 μM)35, compared to the 1000-fold more sensitive M. tuberculosis cytochrome bd knockout strain (MIC ~ 2.0 nM, Fig. 1b).
Finally, we also measured the efficacy of JNJ-2901 in both an acute and a chronic mouse model infected with M. tuberculosis H37Rv-ΔcydAB. In the acute mouse model, treatment with either bedaquiline or JNJ-2901 has started one-week post-infection and continued for two weeks, after which lungs were harvested and plated for bacterial load enumeration (Fig. 1d), resulting in a 3.6-log reduction in bacterial load at an administered dose of 10 mg/kg of JNJ-2901. In the chronic mouse model, treatment was started four weeks post-infection and continued for eight weeks. Here, a 4.0-log reduction in bacterial load was obtained at a dose of 10 mg/kg of JNJ-2901, comparable to the 4.6-log reduction obtained by bedaquiline at 25 mg/kg (Fig. 1e). Hence, JNJ-2901 is an inhibitor of the main mycobacterial proton pump cytochrome bc and can reduce in vivo bacterial loads by four orders of magnitude.
Generation of an M. tuberculosis-like cytochrome bc from M. smegmatis
To characterize the interaction of JNJ-2901 with its target, we aimed to purify the M. tuberculosis cytochrome bc protein complex. However, expression of M. tuberculosis cytochrome bc in M. smegmatis results in low protein yields in our hands. In contrast, M. smegmatis cytochrome bc could be purified in sufficient amounts (1 mg/L of culture) and high purity. Therefore, we set out to create a version of the M. smegmatis cytochrome bc in which the substrate binding site is identical to that of M. tuberculosis. The QcrA and QcrB subunits of cytochrome bc make up the Qp menaquinone active site and share high sequence homology between M. smegmatis and M. tuberculosis (QcrA: 78% identity, QcrB: 82% identity, Supplementary Fig. 1 and 2). Only five amino acid positions in the Qp menaquinone binding site differ between M. smegmatis and M. tuberculosis: Phe156Tyr, Ile182Met, Met189Leu, Asp309Glu, and Ile312Ala (Fig. 2a, Supplementary Fig. 2). Therefore, we engineered a version of M. smegmatis cytochrome bc in which all five residues were mutated to the residues found in M. tuberculosis, which we termed cytochrome bcMtb-like. Next, we purified cytochrome bcMtb-like in large amounts (0.5 mg/L of culture) and homogeneity (Fig. 2b, c) and compared the enzymatic activity of wild-type M. smegmatis cytochrome bc and cytochrome bcMtb-like by an oxygen consumption assay. Here, we find that the oxygen consumption activities of M. smegmatis wild-type cytochrome bc and cytochrome bcMtb-like are identical, indicating that the five mutations do not affect the activity of the enzyme (Fig. 2d). Finally, we measured the impact of JNJ-2901 and that of Q203 on cytochrome bcMtb-like and found that the inhibitory action of both inhibitors on cytochrome bcMtb-like are similar, with IC50 values of 17.4 (±4.8) and 22.4 (±8.4) nM, respectively (Fig. 2e).

a Close-up of the Qp menaquinol binding site of M. smegmatis cytochrome bc. Residues that differ from Mtb cytochrome bc are shown in yellow sticks. See also Supplemental Fig. 2 for sequence alignment. b Chromatogram showing the elution of M. smegmatis cytochrome bcMtb-like on a Superose 6 gelfiltration column. Vertical arrow marks the elution volume of a 670 kDa marker protein (thyroglobulin). c SDS–PAGE showing the purified M. smegmatis cytochrome bcMtb-like. Subunits are indicated to the right of the gel. d Oxygen consumption activity by wild type (blue bar) and Mtb-like (red bar) cytochrome bc. Values were obtained from three separate experiments. Error bars indicate the standard error of the mean. Inset shows an example of oxygen consumption in the presence of cytochrome bc. Vertical arrow marks the addition of the substrate DMQH2. e Inhibitory effect of JNJ-2901 (red curve) and Q203 (blue curve) on the oxygen consumption rate of M. smegmatis cytochrome bcMtb-like. 5 nM protein and 200 µM DMQH2 were used in the assay, with increasing amounts of inhibitor (5 nM–5 µM). Results were obtained from three independent experiments.
Cryo-EM structures of M. smegmatis cytochrome bcMtb-like
To establish how JNJ-2901 interacts with cytochrome bc, we determined the cryo-EM structure of M. smegmatis cytochrome bcMtb-like bound to JNJ-2901 to a resolution of 3.1 Å (Fig. 3, Table 2, Supplementary Fig. 3). Overall, the structure is identical to previous structures of mycobacterial cytochrome bc39. It forms a ‘dimer of dimers’, containing two copies of subcomplex III (bc1) and two copies of subcomplex IV (aa3) (Fig. 3a). Subcomplex III consists of the three subunits: QcrA, QcrB, and QcrC, while subcomplex IV consists of six subunits: CtaC, CtaD, CtaE, CtaF, CtaI, and CtaF, complemented by PRSAF1 (Fig. 3b). At a low map-contour level, the cryo-EM map shows density for the Super Oxide Dismutase (Supplementary Fig. 3e), which also in other publications has been reported to be poorly defined in the cryo-EM map due to its flexibility39. JNJ-2901 is bound in the Qp menaquinol binding site (Fig. 3c), located in the QcrB subunit of the subcomplex III. This is the same binding site as for the other cytochrome bc inhibitors Q203 and TB4724,25. The density for JNJ-2901 is well defined allowing for an accurate positioning of the inhibitor. Only the end of the tail of the inhibitor shows weaker density as it is located out of the ligand binding pocket and has no interaction with the protein. The interactions with the inhibitor are mainly hydrophobic in nature (Fig. 3d) with only two hydrogen bonds between the protein and inhibitor. Almost all contacts are with the QcrB subunit, with a single contact from the QcrA subunit. The five residues that we mutated to make the M. smegmatis ligand binding site identical to M. tuberculosis all interact with JNJ-2901 (marked with a star in Fig. 3d). The cryo-EM map for these residues is well resolved and fits well with the mutated amino acids, but not the original wild type amino acids (Supplementary Fig. 4). Comparison of the position of JNJ-2901 to that of other cytochrome bc inhibitors Q203 and TB47 shows a similar configuration in which the three inhibitors overlap well (Fig. 3e) with the main difference in the tail region, which has few contacts with the protein. The high similarity in binding pose agrees with the similar IC50 values for JNJ-2901 and Q203 on the isolated cytochrome bcMtb-like protein (Fig. 2e).

a Cryo-EM map M. smegmatis cytochrome bcMtb-like bound to JNJ-2901. Subcomplex III (bc1) is shown in light and dark green, subcomplex-IV (aa3) is shown in light and dark blue. PSRAF1 is shown in brown, and the tail of SOD in yellow. b Structure of M. smegmatis cytochrome bcMtb-like shown in cartoon representation. The red square marks the ___location of the JNJ-2901 binding site in one of the two copies of subcomplex III. c Close-up of JNJ-2901 binding site. QcrB is shown in teal, JNJ-2901 in magenta, and cryo-EM map is shown in gray mesh. Underlined residues (His348 & His368) are from the QcrA subunit, all other residues are from the QcrB subunit. d Schematic representation of the interactions between cytochrome bc and JNJ-2901. Hydrophobic interactions are indicated by striated half circles and hydrogen bonds by dashed black lines. Residues mutated to make cytochrome bcMtb-like are marked by a star. e Superimposition of three cytochrome bc inhibitors: JNJ-2901 (magenta), Q203 (gray, PDB:7E1W) and TB47 (sand, PDB:7E1X).
Analysis of JNJ-2901 resistance-inducing mutations
One of the main challenges in the treatment of tuberculosis is the frequent occurrence of resistance against antibiotics1,40,41,42. Also, for the cytochrome bc inhibitors Q203 and TB47, resistance-inducing mutations have been reported17,34,43,44,45,46,47,48,49,50,51,52,53 Therefore, we wanted to determine if M. tuberculosis can also develop resistance against JNJ-2901. For this, M. tuberculosis H37Rv-ΔcydAB was exposed to a high concentration of JNJ-2901 (100 × MIC) and grown for four weeks. After incubation, single colonies were picked and submitted for genome sequencing, revealing twelve mutations that correspond to three amino acid positions located in the ligand binding pocket of cytochrome bc (Table 3, Fig. 4a). Two of these are in the QcrB subunit, QcrBA317V (10×) and QcrBM342T (1×), and one in the QcrA subunit, QcrAL356W (1×). Changes in these amino acid positions have also been observed in laboratory-derived mutations that drive resistance against Q203 and TB4717,23,47,53,54 (Supplementary Table 1). To further characterize the JNJ-2901 resistance-inducing mutations, we created three variants of M. smegmatis cytochrome bcMtb-like: QcrBA312V, QcrBM337V, or QcrAL349W (Table 3, Fig. 4a) and expressed and purified the mutant proteins. Next, we compared the inhibitory effect of JNJ-2901 on the activity of these mutant proteins. We find that all three mutants show a reduced sensitivity to JNJ-2901 (Fig. 4b), with the two mutants in the QcrB subunit showing more than a 700-fold increase in IC50 when compared to the wild-type protein (wild-type: 19 nM, QcrBA312V: 23.8 µM, QcrBM337V: 12.9 µM), while the QcrAL349W mutation showing a modest 5-fold increase in IC50 compared to wild type protein (QcrAL349W: 98 nM). Both QcrBA312 and QcrBM337 are part of the hydrophobic network that interacts with JNJ-2901 (Fig. 3d), while QcrAL349 is located further away (at 4.1 Å) and is positioned next to a cavity that could accommodate the larger sidechain of a tryptophane. This could explain the lesser impact of this mutation on the IC50 of JNJ-2901. These mutant proteins will further serve as tools for the screening of novel inhibitors targeting the resistant enzyme.

a Close-up of the JNJ-2901 binding pocket with JNJ-2901 shown in magenta and mutated residues shown in yellow sticks. b Sensitivity of wild type and JNJ-2901-resistant mutants cytochrome bc protein as measured in an oxygen consumption assay. Reaction mixtures contained 30 nM protein and 200 µM DMQH2, with increasing amounts (5 nM–5 µM) of inhibitor. Results are shown of four independent experiments per protein variant.
Discussion
The widespread prevalence of tuberculosis, combined with an increase in antibiotic-resistant cases, is a major threat to the world’s health. To combat antibiotic resistance, new drugs that inhibit novel targets are needed. The bacterial proton pump cytochrome bc is at the heart of the bacterial respiratory chain, and it is critical for the generation of ATP in bacteria. Novel inhibitors that act on cytochrome bc such as Telacebec (Q203), have passed phase 1 clinical trials and showed promising results in phase 2a20,55, while TB47 is currently in pre-clinical studies. To ensure that a cytochrome bc inhibitor will reach the treatment stage it is important to include new compounds in the drug development pipeline. In this work, we show that JNJ-2901, a novel analog of Q203 and TB47, is a potent growth inhibitor of clinical strains of M. tuberculosis in vitro, with a MIC of less than 1 nM. In addition, JNJ-2901 is effective in both an acute and chronic mouse model infected with M. tuberculosis H37Rv-ΔcydAB, resulting in a 4-log reduction of bacterial load in the lungs, similar to the reduction observed for the widely used bedaquiline.
The cryo-EM structure of cytochrome bc bound to JNJ-2901 shows a canonical binding mode like other inhibitors with an extensive network of hydrophobic interaction. We furthermore show that three resistance-inducing mutations are positioned in the inhibitor binding pocket and that two of these mutations increase the IC50 by >700-fold. It should be noted that these mutations were generated in a laboratory setting and do not necessarily reflect resistance mutations that may occur in a clinical setting. Moreover, current treatment regimens for drug-susceptible and drug-resistant M. tuberculosis include multiple antibiotics to reduce the chances of developing drug resistance. Likewise, we envision that JNJ-2901 should also be used in combination with other antibiotics. Therefore, in a parallel publication, we show that JNJ-2901 is a good partner for the BPaL regimen that is currently used for drug-resistant TB (Aguilar-Perez, submitted, https://doi.org/10.21203/rs.3.rs-5331796/v1) and could be used as a replacement for moxifloxacin in fluoroquinolone-resistant TB. Taken together, our work shows that JNJ-2901 is a promising novel inhibitor of M. tuberculosis cytochrome bc, with the potential of becoming an asset in the ongoing battle against TB. JNJ-2901 should also be further studied as a therapeutic option for Buruli ulcer56, a neglected tropical disease caused by Mycobacterium ulcerans, and Leprosy57 caused by Mycobacterium leprae. Both mycobacteria lack the gene encoding cytochrome bd in their genome, indicating that they rely completely on cytochrome bc for proton pump activity58,59. This is validated by cytochrome bc inhibitors showing promising results in this direction46,60,61.
Methods
Chemicals and DNA primers
All chemicals were purchased from Sigma-Aldrich unless stated otherwise. DNA oligos were purchased from IDT-DNA.
Bacterial strains and growth conditions
The M. tuberculosis H37Rv strain was kindly provided by Roland Brosch (Institut Pasteur, France). To prepare frozen stocks, H37Rv was grown in Middlebrook 7H9 (Becton-Dickinson) culture medium supplemented with 10% oleic acid–albumin–dextrose–catalase (OADC) complex (Becton-Dickinson) and 0.05% Tween 80 (Sigma-Aldrich). Upon reaching the stationary phase, the H37Rv culture was harvested in glycerol (15%) (Becton-Dickinson) containing Middlebrook 7H9 medium (supplemented with 10% OADC and 0.05% Tween 80) and frozen at –80 °C.
Minimal inhibitory concentration determination on clinical isolates of M. tuberculosis
18 M. tuberculosis clinical strains harboring various drug susceptibility profiles were selected from the French NRC of mycobacteria collection (including 13 MDR-TB strains). Drug susceptibility profile was determined by using the BACTEC™ MGIT 960 system for the first-line drug and the reference proportion method in Löwenstein-Jensen for second-line drug.
The agar dilution method was performed on 7H11 supplemented with 10% Middlebrook OADC (oleic acid, albumin, dextrose, and catalase) (BD). Two-fold dilutions of the compounds were added to obtain the final concentrations. A 1/100 dilution of a McFarland 1.0 turbidity standard suspension was inoculated with a Steers replicator delivering approximately 104 CFU per spot. Plates were incubated at 37 °C and MICs were determined after 4 weeks of incubation. The MIC was defined as the lowest concentration of antibiotic resulting in complete inhibition of growth or in the growth of fewer than 10 colonies (<1% of the inoculum).
Minimal inhibitory concentration determination on lab strain M. tuberculosis H37Rv ΔcydAB
Compounds screened in dose-response were tested in 4-fold dilutions from 5 mM to 0.005 µM in black, clear bottom, 384-well microtiter plates (Greiner). Using an Echo liquid handler (Labcyte Inc., Sunnyvale, CA, USA) a low volume dilution range in 100% DMSO was dispensed in the plates (150 nl/well). Reference plates were included as well as positive and negative control wells in each plate. M. tuberculosis H37Rv ΔcydAB (1 × 105 CFU/m) diluted in Middlebrook 7H9 medium supplemented with 10% OADC, 0.5% glycerol, and 0.05% Tween 80 was added to the compound plates (30 µL). Plates were stacked in iron racks and incubated for 5 days at 37 °C. Prior absorbance measurements (OD620) using an Envision multimode plate reader (Perkin Elmer) the pates were shaken (4 min, 1000 rpm). MIC50 values were determined as the drug concentration inhibiting 50% of the growth observed in the control wells.
Generation of JNJ-2901 resistant strains in M. tuberculosis H37Rv ΔcydAB
To isolate resistant colonies, agar plates containing 100 × MIC90 of JNJ-2901 were inoculated with M. tuberculosis H37Rv ΔcydAB (5 × 108 CFU) to select resistant colonies. Individual colonies were re-plated in the presence of the compound for ~3 weeks to confirm resistance. Genomic DNA was isolated from resistant colonies using the Quick-DNA fungal and bacterial miniprep kit (Zymo Research). WGS libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina). Sequencing was performed on an Illumina NextSeq 550 platform, generating paired-end 75 bp reads and targeting 5 M sequences per sample. The trimmed reads were mapped to the reference genome of M. tuberculosis H37Rv (GenBank accession number NC_000962.3) and to the M. tuberculosis H37Rv ΔcydAB control. Variants were identified using the CLC Genomics Workbench v21.0.5 (Qiagen) variant caller, with a minimum count of 2, minimum coverage of 10, and a minimum frequency of 10%. Variants present in all samples and wild-type samples were filtered out. Only variants present in >75% of the reads were considered to minimize noise.
Animal Ethics statement
All the in vivo studies were performed at Johnson & Johnson Innovative Medicines in Beerse, in a certified BSL3 facility in agreement with European Directive (2010/63/EU) and national directives for the protection of animals for experimental purposes. All procedures were approved by the Ethics Committee of Johnson & Johnson Innovative Medicines, located in Beerse, Belgium, which has been accredited by AAALAC since 2004 under unit number 001131 (https://www.aaalac.org/).
Mice
Six to eight weeks old female Balb/cBy mice were purchased from Charles-River (France) or Janvier (France). Mice were allocated in the BLS3 facility in individually ventilated cages in HEPA-filtered racks and rested for at least 5 days to allow acclimatization. Access to water and food was ad libitum. General anesthesia was practiced to infect mice intranasally and for unconscious euthanasia followed by cervical dislocation. Groups of 6 mice were placed in the anesthesia box (integrated gas anesthesia system of the Techniplast Aria Tech60 GAS biosafety cabinet using GE HealthCare Tec 850 vaporizer). Anesthesia was performed using inhalation of isoflurane (Alivira Isoflutek 1000) at 5% for induction and 2.5–3% for maintenance.
Short acute model
The acute mouse model infections were performed as described in refs. 62,63. Mice were infected intranasally with 200 cfu per mouse. To verify the infection level a subgroup of 6 mice were sacrificed one day after the infection. Mice were infected for a week when treatment started, mice were treated daily during 12 consecutive days, mice were euthanized 3 days after the last dose to prevent carry over effect. To control the evolution of the infection, a group of mice was euthanized 7 days post-infection, when treatment starts; and 21 days post-infection, when treatment has ended. Negative control mice remained untreated. Mice were dosed by oral gavage (100 µL, drencher with rounded end straight, 0.9 mm × 25 mm, Socorex Swiss) except for the long-acting formulation that was injected subcutaneously in the upper back (100 µL) using a needle (26Gx13mm, BD MicrolanceTM). At sacrifice, whole lungs are aseptically collected in Gentlemacs tubes (M tubes with strainer, Miltenyi Biotec) containing 2 ml of PBS and homogenized using the RNA_01_01 settings of GentleMACSTM Octo Dissociator (Miltenyi Biotec). Lung homogenate was diluted in PBS and plated in 7H10 charcoal agar plates containing antibiotics (amphotericin: 100 µg/ml, Polymyxin B: 25 µg/ml, Carbenecillin: 50 µg/ml, Trimethoprim: 20 µg/ml 0.5 mg/ml). Plates were incubated at 37 °C during 3–5 weeks. After that, CFU counts were recorded, and data was expressed in the mean log CFU/lung for each group. Statistical analysis was done by one-way ANOVA with Sidak’s test for multiple comparisons in GraphPad Prism.
Chronic model
The chronic mouse model infections were performed as described in ref. 64. Mice were infected intranasally with 200 cfu per mouse. To verify the infection level a subgroup of 6 mice were sacrificed one day after the infection. Mice were infected for a month before treatment started; treatment lasted for 8 weeks. Six mice were sacrificed at the start of treatment as pretreatment control. Negative control mice remained untreated. Mice were dosed by oral gavage (100 µL, drencher with rounded end straight, 0.9 mm × 25 mm, Socorex Swiss) except for the long-acting formulation that was injected subcutaneously in the upper back (100 µL) using a needle (26Gx13mm, BD MicrolanceTM). At sacrifice, whole lungs are aseptically collected in Gentlemacs tubes (M tubes with strainer, Miltenyi Biotec) containing 2 ml of PBS and homogenized using the RNA_01_01 settings of GentleMACSTM Octo Dissociator (Miltenyi Biotec). Lung homogenate was diluted in PBS and plated in 7H10 charcoal agar plates containing antibiotics (amphotericin: 100 µg/ml, Polymyxin B: 25 µg/ml, Carbenecillin: 50 µg/ml, Trimethoprim: 20 µg/ml 0.5 mg/ml). Plates were incubated at 37 °C during 3–5 weeks. After that, CFU counts were recorded, and data were expressed in the mean log CFU/ lung for each group. Statistical analysis was done by one-way ANOVA with Sidak’s test for multiple comparisons in GraphPad Prism.
Construction of a cytochrome bc expression vector
The M. smegmatis qcrCAB operon that encodes for complex III of cytochrome bc was cloned into an acetamide inducible plasmid pACE-GFP-His65 to create the expression plasmid pACEqcrCAB-Ms for purification of M. smegmatis cytochrome bc. To make the M. smegmatis Qp menaquinone substrate binding site identical to that of M. tuberculosis cytochrome bc, five mutations (F156Y, I182M, M189L, D309E, and I112A) were introduced in the qcrB substrate binding site. This resulted in the creation of pACEqcrCABMtb-like, which was used for the expression and purification of M. tuberculosis-like M. smegmatis cytochrome bc (cytochrome bcMtb-like) protein. JNJ-2901-resistant variants of M. smegmatis cytochrome bcMtb-like: QcrBT308A, QcrBA312V, QcrBM337V, and QcrAL349W were created through site-directed mutagenesis. All the primers used for cloning are listed in Supplementary Table 2.
Protein expression and purification
Proteins were expressed in M. smegmatis mc2 15536 in 1xYT media supplemented with 100 mM phosphate, 25 mM sulfate, 50 mM ammonium, 100 mM sodium, 50 mM potassium, 0.5% glycerol, 0.05% glucose, 0.2% alpha-lactose, 0.05% tween-80, and 50 µg/ml of hygromycin. A cell culture at OD600 ~ 1.5 was induced with 0.04% acetamide and grown for 6 h at 37 °C, harvested and flash frozen in liquid nitrogen, and stored at −80 °C.
Cell pellets were resuspended in lysis buffer (25 mM HEPES, pH 7.5, 25 mM imidazole pH 7.5, and 100 mM NaCl) and lysed by passing through an Emulsifex-C5 cell disruptor (Avestin, Inc.). The lysate was spun at 6000 × g to remove the unlysed cells, and then the supernatant was at 257,000 × g for 1 h. The isolated membrane was resuspended in solubilization buffer (25 mM HEPES pH 7.5, 25 mM imidazole pH 7.5 and 100 mM NaCl). Then 1% DDM (n-Dodecyl-ß-d-maltoside) detergent was added and incubated on a rocking platform at 4 °C for 1 h, followed by a centrifugation step at 17,000 × g. The supernatant was loaded onto a Ni-NTA column (Cytiva) and pre-equilibrated in a solubilization buffer with 0.05% DDM. Cytochrome bc was eluted from the column with a gradient of 25–500 mM imidazole in elution buffer (25 mM HEPES pH 7.5, 100 mM NaCl, 0.05% DDM). The protein was concentrated in a 100 kDa centrifugal filter device (Amicon Ultra) and injected onto a pre-equilibrated Superose 6 Increase size exclusion column (GE Healthcare) and run with gel filtration buffer (50 mM HEPES pH 7.5, 100 mM NaCl, and 0.02% DDM). The protein was concentrated to 5 µM, flash frozen in liquid nitrogen, and stored at −80 °C.
Oxygen consumption assay on membrane vesicles
Membrane fractions were prepared from M. smegmatis strains mc2 155 wild type, an M. smegmatis cytochrome bd knockout strain (cydA::hyg), and an M. smegmatis cytochrome bc knockout strain (qcrABC::hyg). Oxygen consumption assays were performed using membrane vesicles with a total of 30 µg of protein and 200 µM NADH in 50 mM HEPES pH 7.5, 100 mM NaCl in a final volume of 600 µl at 37 °C. For the determination of IC50 values, increasing amounts of inhibitor were added from 5 nM to 5 µM.
Oxygen consumption assay on purified protein
Cytochrome bc enzymatic activity was measured in an Oxytherm+ instrument (Hensatech Instrument Ltd.) as described in ref. 24 with a few modifications. The menaquinol mimic 2,3-dimethyl-[1,4]naphthohydroquinone (DMW) (Enamine) was reduced by mixing a 1:10 molar ratio of DMW and DTT for 10 mins at 37 °C to form DMWH2. To start the assay 5 nM of the enzyme, 50 µM of DMWH2, and 50 nM bovine SOD were mixed in 50 mM HEPES pH 7.5, 100 mM NaCl in a final volume of 600 µl and oxygen consumption was measured for 2 mins at room temperature. For the determination of IC50 values, increasing amounts of inhibitor were added from 5 nM to 5 µM.
Cryo-EM sample preparation and imaging
Quantifoil grids (R 2/1, 300 mesh) were glow-discharged using the EMITECH K950 instrument (Quorum) with 20 mA current in 0.2 m Torr vacuum for 45 s. 3 µl of 5 µM purified cytochrome bcMtb-like complex was mixed with JNJ-2901 in a 1:10 molar ratio and applied on the glow discharged grid. The grid was blotted in 85% humidity at 6 °C for 1 s and plunge frozen in liquid ethane using a GP2 instrument (Leica). Cryo-EM data were collected at The Netherlands Center for Electron Nanoscopy (NeCEN) on a Titan Krios (FEI) electron microscope operating at 300 kV with a K3 direct electron detector equipped with a BioQuantum energy filter (Gatan) set to 20 eV. Images were recorded at ×105.000 magnification, 0.836 Å pixel size, and a total dose of 50 e−/Å2, using defocus values from 0.8 to 2.5 µm. Image recording was done in counting mode in EPU software (Thermo Fisher Scientific). The parameters used for image collection are listed in Table 1.
Cryo-EM image processing
Cryo-EM images were processed in Relion 4.066. Images were motion-corrected using Motioncor267 and defocus was estimated using Gctf68. Laplacian-of-Gaussian-based (LoG) autopicking was performed on a subset of micrographs and picked particles were two-dimensionally (2D) classified. Selected classes from the 2D classification were used as references to autopick particles from the full dataset, resulting in 678,771 particles from 4522 micrographs. After three rounds of 2D classification, 192,000 particles derived from classes with different orientations were selected for 3D classification. One of the 3D classes with 42,000 particles was further 3D refined. Defocus values were further refined using CTF Refinement in Relion, followed by Bayesian polishing. Another round of 3D autorefinement was performed on these polished particles. The map was post-processed to correct for the modulation transfer function of the detector and sharpened by applying a negative B factor, as determined automatically by Relion. A soft mask was applied during postprocessing to generate the FSC curve. A starting model of M. smegmatis cytochrome bc (PDB code: 6ADQ22, was fitted to the cryo-EM map using Coot69 and refined in Phenix70. An atomic model for JNJ-2901 was generated using the ligand builder in Coot, and a geometry restraint file was generated in PRODRG71. Figures were prepared using Pymol72, ChimeraX73, and LigPlot+74. Details on data processing, model refinement, and validation are given in Table 1 and Supplementary Fig. 3.
Data availability
The datasets used in this study are available from the corresponding author on request. The cryo-EM map has been submitted to the EMDB under access code: EMD-51689. The molecular model has been submitted to the wwwPDB under access code: 9GY6.
References
WHO. Global Tuberculosis Report 2023 (WHO, Geneva, 2023).
WHO. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-resistant Tuberculosis Treatment, 2022 Update (WHO, 2022).
Andries, K. et al. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS ONE 9, e102135 (2014).
Derendinger, B. et al. Bedaquiline resistance in patients with drug-resistant tuberculosis in Cape Town, South Africa: a retrospective longitudinal cohort study. Lancet Microbe 4, e972–e982 (2023).
Satapathy, P. et al. Emerging bedaquiline resistance: a threat to the global fight against drug-resistant tuberculosis. J. Biosaf. Biosecur. 6, 13–15 (2024).
Nguyen, T. V. A. et al. Bedaquiline resistance: its emergence, mechanism, and prevention. Clin. Infect. Dis. 66, 1625–1630 (2018).
WHO. CALL TO ACTION: Shorter and More Effective Treatment for all People Suffering from Drug-resistant TB (WHO, 2023).
Gupta, A. et al. Global adoption of 6-month drug-resistant TB regimens: projected uptake by 2026. PLoS ONE 19, e0296448 (2024).
Saluzzo, F., Adepoju, V. A., Duarte, R., Lange, C. & Phillips, P. P. J. Treatment-shortening regimens for tuberculosis: updates and future priorities. Breathe 19, 230028 (2023).
Conradie, F. et al. Treatment of highly drug-resistant pulmonary tuberculosis. N. Engl. J. Med. 382, 893–902 (2020).
Preiss, L. et al. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 1, e1500106 (2015).
Koul, A. et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3, 323–324 (2007).
Andries, K. et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science (New York, NY) 307, 223 (2005).
Bald, D., Villellas, C., Lu, P. & Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 8, https://doi.org/10.1128/mbio.00272-17 (2017)..
Cook, G. M. et al. Oxidative phosphorylation as a target space for tuberculosis: success, caution, and future directions. Microbiol. Spectr. 5, https://doi.org/10.1128/microbiolspec.tbtb2-0014-2016 (2017).
Foo, C. S.-Y., Pethe, K. & Lupien, A. Oxidative phosphorylation—an update on a new, essential target space for drug discovery in Mycobacterium tuberculosis. Appl. Sci. 10, 2339 (2020).
Pethe, K. et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19, 1157–1160 (2013).
Lu, X. et al. Pyrazolo[1,5-a]pyridine Inhibitor of the respiratory cytochrome bcc complex for the treatment of drug-resistant tuberculosis. ACS Infect. Dis. 5, 239–249 (2019).
Kim, J. et al. Safety, Tolerability, pharmacokinetics, and metabolism of telacebec (Q203) for the treatment of tuberculosis: a randomized, placebo-controlled, multiple ascending dose phase 1B trial. Antimicrob. Agents Chemother. 67, e01123-22 (2023).
de Jager, V. R. et al. Telacebec (Q203), a new antituberculosis agent. N. Engl. J. Med. 382, 1280–1281 (2020).
Trumpower, B. L. Cytochrome bc1 complexes of microorganisms. Microbiol. Rev. 54, 101–129 (1990).
Gong, H. et al. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. Science 362, eaat8923 (2018).
Wiseman, B. et al. Structure of a functional obligate complex III2IV2 respiratory supercomplex from Mycobacterium smegmatis. Nat. Struct. Mol. Biol. 25, 1128–1136 (2018).
Yanofsky, D. J. et al. Structure of mycobacterial CIII2CIV2 respiratory supercomplex bound to the tuberculosis drug candidate telacebec (Q203). eLife 10, e71959 (2021).
Zhou, S. et al. Structure of Mycobacterium tuberculosis cytochrome bcc in complex with Q203 and TB47, two anti-TB drug candidates. eLife 10, e69418 (2021).
Bosch, B. et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 184, 4579–4592.e4524 (2021).
da Silva, F. F. et al. Unveiling the antimycobacterial potential of novel 4-alkoxyquinolines: insights into selectivity, mechanism of action, and in vivo exposure. J. Med. Chem. 67, 21781–21794 (2024).
Borsoi, A. F. et al. Exploring scaffold hopping for novel 2-(quinolin-4-yloxy)acetamides with enhanced antimycobacterial activity. ACS Med. Chem. Lett. 15, 493–500 (2024).
Murnane, R. et al. Synthesis and antitubercular activity of novel 4-arylalkyl substituted thio-, oxy- and sulfoxy-quinoline analogues targeting the cytochrome bc1 complex. Bioorg. Chem. 138, 106659 (2023).
Liu, R. et al. Syntheses and studies of deuterated Imdiazo[1,2-a]pyridine-3-carboxamides with potent anti-tuberculosis activity and improved metabolic properties. Bioorg. Chem. 128, 106074 (2022).
Wani, M. A. & Dhaked, D. K. Targeting the cytochrome bc1 complex for drug development in M. tuberculosis: review. Mol. Divers. 26, 2949–2965 (2022).
Bajeli, S. et al. Terminal respiratory oxidases: a targetables vulnerability of mycobacterial bioenergetics? Front. Cell. Infect. Microbiol. 10 (2020).
Kalia, N. P. et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl Acad. Sci. USA 114, 7426–7431 (2017).
Arora, K. et al. Respiratory flexibility in response to inhibition of cytochrome C oxidase in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 6962–6965 (2014).
Lu, P. et al. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci. Rep. 8, 2625 (2018).
Snapper, S. B. et al. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4, 1911–1919 (1990).
Matsoso, L. G. et al. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 187, 6300–6308 (2005).
Kana, B. D. et al. Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J. Bacteriol. 183, 7076–7086 (2001).
Mathiyazakan, V., Wong, C.-F., Harikishore, A., Pethe, K. & Grüber, G. Cryo-electron microscopy structure of the Mycobacterium tuberculosis cytochrome bcc-aa3 supercomplex and a novel inhibitor targeting subunit cytochrome ci. Antimicrob. Agents Chemother. 67, e01531–01522 (2023).
Dartois, V. A. & Rubin, E. J. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat. Rev. Microbiol. 20, 685–701 (2022).
Lange, C. et al. Management of drug-resistant tuberculosis. Lancet 394, 953–966 (2019).
Seung, K. J., Keshavjee, S. & Rich, M. L. Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis. Cold Spring Harbor Perspect. Med. 5, a017863 (2015).
Chandrasekera, N. S. et al. Improved phenoxyalkylbenzimidazoles with activity against Mycobacterium tuberculosis appear to target QcrB. ACS Infect. Dis. 3, 898–916 (2017).
Cleghorn, L. A. T. et al. Identification of morpholino thiophenes as novel Mycobacterium tuberculosis inhibitors, targeting QcrB. J. Med. Chem. 61, 6592–6608 (2018).
Foo, C. S. et al. Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of Mycobacterium tuberculosis. mBio 9, https://doi.org/10.1128/mbio.01276-18 (2018)..
Gao, Y. et al. Ultra-short-course and intermittent TB47-containing oral regimens produce stable cure against Buruli ulcer in a murine model and prevent the emergence of resistance for Mycobacterium ulcerans. Acta Pharm. Sin. B 11, 738–749 (2021).
Gries, R. et al. Characterization of two novel inhibitors of the Mycobacterium tuberculosis cytochrome bc(1) complex. Antimicrob. Agents Chemother. 67, e0025123 (2023).
Harrison, G. A. et al. Identification of 4-amino-thieno[2,3-d]pyrimidines as QcrB inhibitors in Mycobacterium tuberculosis. mSphere 4, https://doi.org/10.1128/msphere.00606-19 (2019)..
Lupien, A. et al. New 2-ethylthio-4-methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLoS Pathog. 16, e1008270 (2020).
Phummarin, N. et al. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc1 inhibitors. MedChemComm 7, 2122–2127 (2016).
Rybniker, J. et al. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 6, 7659 (2015).
van der Westhuyzen, R. et al. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: a novel antimycobacterial class targeting mycobacterial respiration. J. Med. Chem. 58, 9371–9381 (2015).
Waller, N. J. E., Cheung, C.-Y., Cook, G. M. & McNeil, M. B. The evolution of antibiotic resistance is associated with collateral drug phenotypes in Mycobacterium tuberculosis. Nat. Commun. 14, 1517 (2023).
Moraski, G. C. et al. Arrival of Imidazo[2,1-b]thiazole-5-carboxamides: potent anti-tuberculosis agents that target QcrB. ACS Infect. Dis. 2, 393–398 (2016).
Kim, J. et al. Safety, tolerability, and pharmacokinetics of telacebec (Q203), a new antituberculosis agent, in healthy subjects. Antimicrob. Agents Chemother. 66, e01436–01421 (2022).
Demangel, C., Stinear, T. P. & Cole, S. T. Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat. Rev. Microbiol. 7, 50 (2009).
Gilmore, A., Roller, J. & Dyer, J. A. Leprosy (Hansen’s disease): an update and review. MO Med. 120, 39–44 (2023).
Scherr, N. et al. Targeting the Mycobacterium ulcerans cytochrome bc1:aa3 for the treatment of Buruli ulcer. Nat. Commun. 9, 5370 (2018).
Cole, S. T. et al. Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011 (2001).
Liu, Y. et al. The compound TB47 is highly bactericidal against Mycobacterium ulcerans in a Buruli ulcer mouse model. Nat. Commun. 10, 524 (2019).
Converse, P. J., Almeida, D. V., Tyagi, S., Xu, J. & Nuermberger, E. L. Shortening Buruli ulcer treatment with combination therapy targeting the respiratory chain and exploiting Mycobacterium ulcerans gene decay. Antimicrob. Agents Chemother. 63, https://doi.org/10.1128/aac.00426-19 (2019)..
Ray, P. C. et al. Spirocycle MmpL3 inhibitors with improved hERG and cytotoxicity profiles as inhibitors of Mycobacterium tuberculosis growth. ACS Omega 6, 2284–2311 (2021).
Akester, J. N. et al. Synthesis, structure–activity relationship, and mechanistic studies of aminoquinazolinones displaying antimycobacterial activity. ACS Infect. Dis. 6, 1951–1964 (2020).
Lenaerts, A. J. et al. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 49, 2294–2301 (2005).
Arnold, F. M. et al. A uniform cloning platform for mycobacterial genetics and protein production. Sci. Rep. 8, 9539 (2018).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Elife 11, e83724 (2022).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Schüttelkopf, A. W. & Van Aalten, D. M. F. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 1355–1363 (2004).
The PyMOL molecular graphics system, Version 2.5, Schrödinger, LLC.
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Acknowledgements
We thank Valerie Mizrahi for providing M. smegmatis cytochrome BC and cytochrome BD knockout strains and Dirk Bald and Lars J.C. Jeuken for their help with the oxygen consumption assay. This work has received support from the EU/Janssen Pharmaceutica Innovative Medicines Initiative 2 Joint Undertaking RespiriTB grant no. 853903. We thank the staff of the LUMC EM facility and The Netherlands Center for Electron Nanoscopy (NeCEN) for help with data collection and data processing. Access to NeCEN was supported by the Netherlands Electron Microscopy Infrastructure (NEMI), project 184.034.014 of the National Roadmap for Large-Scale Research Infrastructure of the Dutch Research Council (NWO).
Author information
Authors and Affiliations
Contributions
M.H.L. and D.A.L. conceived and supervised the study. A.K.V. purified the M. smegmatis Mtb-like cytochrome bc1:aa3 complex, determined the cryo-EM structure, and built the structural model. A.K.V. and R.Q.K. performed enzymatic on purified protein. A.A. and N.V. designed and supervised MIC studies on clinical isolates of M. tuberculosis. S.W. performed MIC studies on clinical isolates of M. tuberculosis. J.W. and C.A.P. performed mouse studies. M.H.L., D.A.L., J.M.B., and R.J.C. analyzed the structure. The manuscript was written by M.H.L. and A.K.V. with help from all authors. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.K.V., R.Q.K., M.H.L., S.W., N.V., and A.B. have no competing interests. D.L., C.A.P., J.M.B., R.J.C., and J.W. are full-time employees of Janssen, a Johnson & Johnson company, and/or potential stockholders of Johnson & Johnson. J.G., C.V., and D.A.L. have been named inventors in a patent application for JNJ-2901.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verma, A.K., Kim, R.Q., Lamprecht, D.A. et al. Structural and mechanistic study of a novel inhibitor analogue of M. tuberculosis cytochrome bc1:aa3. npj Drug Discov. 2, 6 (2025). https://doi.org/10.1038/s44386-025-00008-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44386-025-00008-3
