
Abstract
Follicular thyroid carcinoma (FTC) is a common endocrine malignancy characterized by a higher propensity for invasion and metastasis compared to papillary thyroid carcinoma (PTC). Ephrin type A receptor 5 (EPHA5) is a crucial receptor tyrosine kinase involved in orchestrating diverse physiological processes, including apoptosis and proliferation. However, the mechanism of EPHA5 in FTC remains unclear. This study identified significant overexpression of EPHA5 in FTC. In vitro experiments showed that increased expression of EPHA5 promotes proliferation and inhibits apoptosis in FTC. Furthermore, EPHA5 activates the STAT3 signaling pathway. To explore the interaction between EPHA5 and the STAT3 signaling pathway, we used SH-4-54 (a STAT3-specific inhibitor). Interestingly, the influence of EPHA5 on proliferation and apoptosis was reduced upon combination with SH-4-54. In summary, this study unveils the involvement of the EPHA5-STAT3 signaling pathway in FTC and implies that the function of EPHA5 in FTC may partly depend on the STAT3 signaling pathway.

Similar content being viewed by others

Introduction
Thyroid cancer (TC) is the most common endocrine tumor. Data from the National Cancer Center of China spanning 2000 to 2016 indicates that thyroid cancer constitutes 4.98% of all newly diagnosed cancers, with a noteworthy annual rise in incidence among women [1]. In contrast to trends observed in Western countries, a statistical analysis of thyroid cancer in the United States from 2000 to 2018 reveals a decrease in incidence but a gradual rise in mortality rates [2]. The treatment approach for thyroid cancer emphasizes early detection, timely diagnosis, and prompt surgical intervention. However, when surgical intervention is not feasible, alternatives like radiotherapy or gene therapy may be considered. More than 90% of TC is represented by differentiated TC (DTC) that arises from the follicular thyroid cells. DTC includes papillary TC (PTC), follicular TC (FTC), and Hürthle cell TC. Anaplastic TC (ATC) accounts for 1% of TC, and it represents 15–40% of TC-related deaths. Current treatment strategies are not completely effective against aggressive DTC or ATC, and mortality remains one of the most important challenges [3]. FTC is highly malignant and presents challenges for clinical diagnosis. Additionally, a subset of patients may develop metastasis during extended follow-up periods. Furthermore, follicular thyroid carcinoma is associated with RAS mutations, and there are currently no targeted therapeutic drugs available demonstrating efficacy. Follicular thyroid carcinoma is characterized by high malignancy and poses challenges for clinical diagnosis. Active investigation into the precise pathogenesis of follicular thyroid cancer and the search for viable gene therapy targets may emerge as the future frontier in its treatment [4].
Eph receptors (EPHs) make up the largest family of Receptor Tyrosine Kinases (RTKs), with 14 members distinguished into two types, A and B, dependent on sequence similarity and their preferential binding to type A and type B ephrin ligands, respectively. Initial investigations into EPH genes in humans initially focused on oncology, uncovering significantly elevated tissue expression in various tumors, such as hepatocellular cancer, prostate cancer, and ovarian cancer, among others [5,6,7]. Depending on the cellular context, these diverse signaling modalities can induce varied, and occasionally contrasting, effects on cancer cells. This variability stems from mechanisms such as chromosome amplification or loss, transcriptional regulation of oncogenic signaling pathways, promoter methylation, and the influence of microRNAs [6, 8, 9]. Furthermore, members of the EPH family and their ligands exhibit significant efficacy in regulating tumor cell growth, invasion, metastasis, and neovascularization [6, 10,11,12,13]. Notably, our previous experiments utilizing next-generation sequencing technology revealed a significant up-regulation of EPHA5 expression in FTC tissues compared to their correspondingx paired para-carcinoma tissues. Interestingly, EPHA5 possesses tyrosine kinase activity and serves as a signal transduction receptor. Based on previous evidence, we hypothesize that EPHA5 potentially plays a pivotal role in the development of follicular thyroid carcinoma.
The STAT (Signal Transducer and Activator of Transcription) protein family encompasses various transcription factors crucial for regulating diverse cellular physiological processes, including proliferation, differentiation, apoptosis, angiogenesis, and mechanisms pertaining to immune regulation. As a member of the STAT family, STAT3 has been extensively shown to play a pivotal role in cancer biology [14,15,16,17,18]. Presently, it is widely regarded as a “ubiquitous” proto-oncogene, with aberrant activation of STAT3 observed in over 70% of human cancers [17, 18]. Additionally, the overexpression of phosphorylated STAT3 (p-STAT3) is markedly correlated with unfavorable clinical prognosis in the majority of tumors [19,20,21,22,23,24]. Thus, the aberrant activation of STAT3 is intricately linked to the biological behavior of tumors [25]. Considering that EPHA5 belongs to the tyrosine kinase family, it may be associated with STAT3. In this study, we found that an inter-binding relationship between EPHA5 and STAT3, indicating that EPHA5 might potentially activate STAT3. However, the precise mechanism underlying the interaction between EPHA5 and STAT3 remains elusive.
The analysis of next-generation sequencing technology results in this study has uncovered a significant increase in EPHA5 expression among patients with FTC. Additionally, the functional impact of EPHA5 on the proliferation and apoptosis of FTC was investigated both in vitro and in vivo. This study has the potential to advance our understanding of the biological properties of EPHA5 tyrosine kinase and may offer a promising target for therapeutic intervention in FTC.
Materials and methods
Next-generation sequencing
The next generation sequencing was conducted by Novogene in Beijing, China. The samples were obtained from the Second Affiliated Hospital of Dalian Medical University. Each sample used 3 mg of RNA for RNA sample preparation. A 1% agarose gel was used to assess RNA degradation and contamination. The Qubit 2.0 fluorometer with the Qubit RNA analysis kit was utilized to quantify RNA concentrations, while the Bioanalyzer 2100 system with the RNA Nano 6000 assay kit was employed to evaluate RNA integrity. The BallCross suite includes interactive decomposition of transcriptome assembly, visualization of transcriptional structures, unique rich features for each locus, and post hoc annotation from assembly features to annotation features. Transcripts with adjusted p values below 0.05 were considered differentially expressed. Each group consisted of three tissue samples.
Tissue collection
FTC specimens were obtained from three patients who provided informed consent and agreed to participate in accordance with the ethical standards of the Second Affiliated Hospital of Dalian Medical University (Dalian, China). Adjacent normal thyroid tissue samples were collected from the same patients, with a distance of more than 2 cm from the tumor lesion. All samples were confirmed by pathologists as FTC based on histopathological evaluation. These patients had not received any local or systemic treatment prior to surgery.
IHC staining analysis
Tumor samples from 20 patients diagnosed with follicular thyroid carcinoma, along with their corresponding adjacent tissues, were fixed in paraffin, sectioned, and stained using hematoxylin and eosin (Clinical and pathological information of patients can be found in Supplementary Table 2). The paraffin sections were then subjected to heat at 60 °C for 30 min. After dewaxing, antigen retrieval was performed using sodium citrate. Subsequently, the sections were treated with 3% H2O2 and blocked with goat serum. Following this, the sections were exposed to anti-EPHA5 antibody (diluted at 1:150), washed with PBS, incubated with biotinylated secondary antibody for 10 min at room temperature. Signal amplification was achieved by treating the sections with 3,3-diaminobenzidine tetrahydrochloride (DAB) followed by counterstaining with hematoxylin for 5 s. Finally, the slides were dehydrated using a series of alcohol gradients before being covered with coverslips and examined under a microscope. The immunostaining intensity was classed into four categories: negative (value = 0), weak (value = 1), moderate (value = 2) and strong (value = 3). The percentage of positive cell was classed into four categories: 0–25% (value = 0), 26–50% (value = 1), 51–75% (value = 2) and 76–100% (value = 3).
Cell culture
The human thyroid cell lines Nthy-ori 3-1, FTC133 and FTC238 were obtained from SHANGHAI WHELAB BIOSCIENCE LIMITED (Shanghai, China). Nthy-ori 3-1 and FTC133 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco) and 1% penicillin-streptomycin solution (Solarbio, Beijing, China). FTC238 cells were cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS and 1% penicillin-streptomycin solution (Solarbio). All cells were maintained in an incubator at 37 °C and 5% CO2.
Cell viability assay
Cells in the logarithmic growth phase will be treated with trypsin and then evenly distributed into 96-well plates. Each set of wells will consist of five replicates. Fill the outer wells of the plate with PBS and place it in a 37 °C, 5% CO2 incubator. Inoculate each well with a total of 5 × 103 cells and allow them to adhere together. After aspirating the medium, treat the cells with specified concentrations of substances for a specific period. Add CCK-8 solution at a rate of 10 ml per well and then incubate the plate at 37 °C for 1 h. Measure the optical density (OD) at 490 nm using a microplate reader for each well, taking an average value, and repeating the experiment at least three times.
Colony formation assay
A total of 1000 cells were mixed well with 3 mL of culture medium containing 10% FBS in six-well plates. The cells were then maintained in an incubator at 37 °C and 5% CO2 for 1 week. After that, the plates were removed, the medium was discarded, and the residual medium was gently washed away with running water. The cells in the wells were fixed with paraformaldehyde for 30 min and then stained with 0.1% crystal violet. Clusters containing 30 or more cells were counted as one colony.
RNA interference and plasmid constructs
siRNA targeting EPHA5, negative control siRNA, and overexpression of EPHA plasmid were purchased from Gene Pharma (Shanghai, China). The sequences of the si-EPHA5 constructs were (si-EPHA5#2: 5’-GCA UGU GCA AGG CAG GAU ATT-3’, and si-EPHA5#3: 5’-CCA GCU ACA CGA UUA UCA ATT-3’). The sequences of the si-STAT3 constructs were (siRNA -STAT3: 5’-UGCUGUAGCUGAUUCCAUUGGGCCA-3’). The transfection procedures were performed according to the protocols. The FTC cells were transfected with specific siRNAs and overexpression of EPHA5 plasmid vectors using Lipofectamine 3000 transfection reagent (Invitrogen, Shanghai, China).
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted using RNAiso Plus (TaKaRa, Kusatsu, Japan), and cDNA synthesis and RNA amplification were performed using Prime Script RT kit and SYBR Premix Ex Taq II (TaKaRa), respectively. The expression level normalized to β-actin was determined in each sample by calculating ΔΔCt. The sequences of the primers used are shown in Supplementary Table 1.
Western blot analysis
Reagents used for western blot analysis included primary antibodies against EPHA5 (obtained from Santa Cruz, USA), STAT3 (purchased from Santa Cruz, USA), p-STAT3 (sourced from Santa Cruz, USA), cleaved caspase-3 (acquired from Beyotime Institute of Biotechnology, Shanghai, China), PCNA (obtained from Proteintech, Wuhan, China), Cyclin D1 (sourced from Proteintech, Wuhan, China) and β-actin (acquired from Beyotime Institute of Biotechnology, Shanghai, China). Protein quantification was conducted using Gel-Pro Analyzer version 4.0 software (Media Cybernetics, MD, USA).
Coimmunoprecipitation (coIP)
Total protein in FTC-238 was extracted using IP lysis buffer (20 mM Tris-HCl, 150 mM NaCl, and 1% Triton X-100, pH 7.5). For coIP experiments, the protein AG magnetic beads (Bimake, Selleck Chemical, Houston, TX, USA) were incubated with amounts of anti-STAT3 antibodies or anti-EPHA5 antibodies and according to the manufacturer’s instructions. The precipitate was washed there times with washing buffer. The pre-extracted protein supernatant and the antibody-bound magnetic beads were incubated at room temperature for 2–3 h. The precipitate was washed there times with washing buffer. Then, 50 ul of 1 × loading buffer was added to resuspend the precipitate, and boiled in boiling water for 10 min. Finally, the supernatant was collected by magnetic separation and western blotted with anti-EPHA5 and anti-STAT3 antibodies together as primary antibody at 4 °C overnight, followed by incubation with an anti-IgG light chain specific secondary antibody (Abbkine Scientific Co., Ltd., USA).
Immunofluorescence assay (IF)
The FTC-238 cells were grown on glass coverslips in 24-well plates, fixed with a 4% solution of formaldehyde, and permeabilized using 0.5% Triton X-100/PBS. After that, the cells were incubated with 5% BSA for half an hour at room temperature and then exposed to primary antibodies against EPHA5 (1:200, Santa Cruz, USA) and STAT3 (1:500, Proteintech, Wuhan, China) overnight at 4 °C. Following this step, they were treated with fluorescent dye-conjugated secondary antibody (Proteintech, Wuhan, China) for 2 h at 37 °C before being mounted with DAPI.
TUNEL assay
FTC cells were plated in 24-well dishes, fixed with a 4% formaldehyde solution, and permeabilized with 0.1% Triton X-100/PBS. Subsequently, the cells were rinsed three times for 5 min each with PBS, followed by the preparation of a fluorescent working solution according to the provided instructions and its addition to each well (50 μ/well). The cells were then incubated in a 37 °C water bath for 1 h. After gently washing with PBS three times for 5 min each, DAPI dye was introduced into each well and incubated in a 37 °C water bath for 30 min. Finally, the cells underwent another round of gentle washing with PBS three times for 5 min each before being observed under a fluorescence microscope.
Annexin V-FITC apoptosis detection kit
FTC cells were grown in 24-well plates, then the cell culture medium was removed and the cells were washed once with PBS. After that, the PBS was centrifuged at 1000 × g for 5 min. Following this, 195 μl of Annexin V-FITC binding solution was added, along with 5 μl of Annexin V-FITC, and gently mixed. Then, 10 μl of propidium iodide staining solution was added and gently mixed again. The mixture was then incubated in the dark at room temperature (20–25 °C) for 10–20 min before being placed on ice; aluminum foil can be used to protect it from light. Finally, it was immediately observed under a fluorescence microscope.
BeyoClick™ EdU-488 cell proliferation test kit
FTC cells were grown in a 24-well plate. EdU was mixed with cell culture medium at a ratio of 1:500 (10 mM) to make a working solution of 20 μM EdU. After warming the 20 μM EdU at 37 °C, an equal amount was added to the wells, resulting in a final concentration of 10 μM EdU in the plate. The cells were then allowed to incubate for an additional 2 h. Once the EdU labeling was complete, the culture medium was replaced with 0.5 ml of 4% paraformaldehyde for fixation at room temperature for 15 min. The fixative was then discarded, and each well was rinsed with washing solution. After removing the washing solution, each well received an addition of 0.5 ml PBS containing 0.3% Triton X-100 and was left at room temperature for approximately 10–15 min to facilitate permeabilization. The permeabilization solution was then removed, followed by one or two washes with washing solution. Next, add Click reaction mixture per well and gently agitate the culture plate to ensure even coverage over samples during incubation away from light at room temperature for 30 min. Subsequently, the Click reaction mixture should be removed and subjected to 3 washes using washing solution. After that, Hoechst staining solution (1×) should be added at a volume of 0.4 ml per well and incubated under light-protected conditions at room temperature for 10 min before removing excess Hoechst stain. The samples should then be thoroughly washed three more times with washing liquid as previously described before being observed under an inverted fluorescence microscope.
Drug treatment
The STAT3 inhibitor (SH-4-54) was purchased from MedChemExpress. For in vivo experiments, SH-4-54 dissolved in DMSO was administered through intraperitoneal injection at a dose of 5 mg/kg every 3 days. For in vitro studies, SH-4-54 was dissolved in sterile DMSO and used to treat FTC cells at a concentration of 10 μM for a duration of 48 h.
Animals
Athymic nude mice (6 weeks old) were procured from the Specific Pathogen-Free (SPF) Animal Experimental Center of Dalian Medical University. The animal care and experimental procedures were reviewed and approved by the Institutional Animal Care and Ethics Committee of Dalian Medical University. After 1 week of adaptation, 20 nude mice were divided into 4 groups marked as (NS group); (NS group + SH-4-54); (group EPHA5 + SH-4-54); or (group si-EPHA5 + SH-4-54) in the right groin (1 × 107 cells in 200 μl of PBS). After 1 week, the tumors had grown to a measurable size. Tumor length (L) and width (W) were measured weekly with calipers, and tumor volume (V) was calculated using the formula V = LW2/2. Nude mice were sacrificed 4 weeks after tumor injection. All tumors were collected within 20 min of euthanasia and promptly frozen in liquid nitrogen.
Statistical analysis
All measurement data are processed with mean ± SD. Statistical analysis software used SPSS 19.0 for data analysis. For the data conforming to the normal distribution, the statistical method of one-way ANOVA was used to compare the mean between groups, and the Turky method was used to test. For the data that does not conform to the normal distribution, the statistical test method adopts the rank sum test (nonparametric test). p < 0.05 indicates statistical difference.
Results
EPHA5 is aberrantly highly expressed in FTC patients
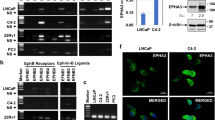
To further investigate the role of the EPH family in follicular thyroid carcinoma, we performed next-generation sequencing on both cancerous tissues and their corresponding paracancerous counterparts from patients diagnosed with follicular thyroid carcinoma. The sequencing results revealed a significant increase in mRNA levels of EPHA5 within the EPH family in follicular thyroid carcinoma tissues compared to paracancerous tissues (Fig. 1A). Additionally, we validated the reliability of our sequencing results by analyzing the datasets of GEO (https://www.ncbi.nlm.nih.gov/geo/): GSE2715 [26, 27], confirming an increased expression of EPHA5 in follicular thyroid carcinoma (Fig. 1B). Furthermore, we assessed the expression levels of EPHA5 in three distinct cell lines: normal thyroid cells (Nthy-ori 3-1) and two follicular thyroid carcinoma cell lines (FTC-133, FTC-238). The EPHA5 mRNA was increased in FTC tissues (Fig. 1C). Additionally, the expression of EPHA5 showed a significant increase in both thyroid follicular carcinoma cell lines compared to the normal thyroid cell lines (Fig. 1D). Furthermore, when comparing the Immunohistochemistry (IHC) results of clinical follicular thyroid carcinoma tissues with their corresponding paraneoplastic tissues, the expression levels of EPHA5 were classified into four categories based on the combination of immunostaining intensity and percentage of positive cells: negative (Score 0–1), weak (Score 2–4), moderate (Score 5–7), and strong (Score 8–12) (Fig. 1E and Supplementary Fig. 1). Based on the preceding results, we concluded that the expression level of EPHA5 was abnormally elevated in FTC. We found that high expression of EPHA5 significantly correlated with advanced clinical stage (Supplementary Table 3). In conclusion, these findings strongly suggest that the abnormal elevation of EPHA5 expression may be implicated in the pathogenesis of follicular thyroid carcinoma.

A The heatmaps and volcano plot show that EPHA5 expression was upregulated in FTC tissues. B Box plot analysis of EPHA5 expression differences. The data are sourced from the datasets of GEO (https://www.ncbi.nlm.nih.gov/geo/): GSE27155. C qPCR analysis of EPHA5 mRNA between Nthy-ori-3-1 and FTC cells, *p < 0.05. n = 6. D Western blot analysis of EPHA5 protein expression in FTC-133 and FTC-238 cells. *p < 0.05. n = 3. E Representative images from immunohistochemically staining of EPHA5 in FTC patient tissues and matching paraneoplastic tissues (EPHA5 is localized in the cell membrane and endoplasmic reticulum). scale bars, 50 μm. n = 20.
Knockdown of EPHA5 inhibits thyroid follicular carcinoma cell proliferation and promotes apoptosis
There is literature indicating that EPHA5 is related to the proliferation of tumor cells [28, 29]. To investigate the impact of EPHA5 on the proliferation and apoptosis of FTC cells, we designed three siRNAs and transfected them into both FTC-133 and FTC-238 cell lines to suppress EPHA5 expression. The efficiency of these three siRNAs was assessed, and the two siRNAs demonstrating higher efficacy were chosen. Following this, we transfected the two selected siRNAs into both FTC-133 and FTC-238 cell lines, and the knockdown efficiency was demonstrated (Fig. 2A). Additionally, we evaluated changes in the expression levels of PCNA, Cyclin D1, and activated Caspase-3 after EPHA5 knockdown, while the protein level of Caspase-3 remained relatively unchanged. Specifically, the levels of PCNA and Cyclin D1 were downregulated, while the levels of activated Caspase-3 were upregulated (Fig. 2A). We then assessed cell proliferation at 24-, 48-, and 72-h post-transfection, as well as the cell survival rate at 24 h post-transfection using CCK-8. As illustrated in Fig. 2B, C, a significant inhibition in cell survival was observed following the suppression of EPHA5 expression. Moreover, the apoptosis rate experienced a significant increase when EPHA5 was inhibited (Fig. 2D), suggesting that the knockdown of EPHA5 indeed promoted apoptosis. Furthermore, cell cloning assays confirmed that the inhibition of EPHA5 expression suppressed proliferation (Fig. 2E). Additionally, after knocking down EPHA5, some FTC cells exhibit impaired spread ability in terms of morphology, which may be related to apoptosis (Supplementary Fig. 2). Transfection of si-EPHA5 in FTC cells, followed by treatment with the apoptosis inhibitor Z-VAD (Z-VAD-FMK), reversed the anti-proliferative effect induced by si-EPHA5 (Supplementary Fig. 3). The EdU incorporation assay and Annexin V/PI staining were further used to confirm that EPHA5 promotes FTC cell proliferation and inhibits apoptosis (Supplementary Figs. 4 and 5).

A Western blot analysis of EPHA5, PCNA, Cyclin D1, Caspase-3, Cleaved-caspase-3, and β-actin in FTC-133 and FTC-238 cells. **p < 0.01, n = 3. B, C CCK-8 assay analysis of FTC-133 and FTC-238 cells transfected with si-NC or si-EPHA5 respectively. **p < 0.01, n = 6. D Representative TUNEL staining of FTC cells; the nuclei were stained with DAPI. Scale bar, 200 μm. TUNEL scores. The relative number of apoptotic cells is represented as TUNEL-positive cells/DAPI. **p < 0.01, n = 6. E Colony formation assay of FTC-133 and FTC-238 cells transfected with si-NC or si-EPHA5 respectively. **p < 0.01, n = 6.
Therefore, our findings strongly support the notion that the suppression of EPHA5 expression leads to the inhibition of FTC cell proliferation and the promotion of apoptosis.
EPHA5 binds to STAT3 and activates STAT3 pathway
Previous experimental results have indeed demonstrated that EPHA5 can influence the proliferation and apoptosis of FTC cells. However, the exact regulatory mechanism governing EPHA5 remains unclear. We screened for proteins that bind to EPHA5 (Fig. 3A), presenting the results from the high probability database, which indicate a binding association between EPHA5 and STAT3. As a member of EPHs, EPHA5 also possesses tyrosine kinase activity. To further investigate the potential mechanism of EPHA5, it was observed that EPHA5 formed a complex with STAT3 through immunoprecipitation, suggesting a binding relationship between EPHA5 and STAT3 (Fig. 3B). The immunofluorescence assay was used to further confirm the binding of EPHA5 with STAT3 (Fig. 3C). The Coip result of EPHA5 and other proteins shows that EPHA5 has a higher binding force with STAT3 (Supplementary Fig. 5). Subsequently, inhibiting EPHA5 expression resulted in a decrease in the phosphorylation level of STAT3 (p-STAT3), while the protein level of STAT3 remained relatively unchanged (Fig. 3D). Additionally, we used the Real-time PCR method to investigate whether inhibiting EPHA5 expression could affect the expression levels of downstream genes associated with STAT3. As shown in Fig. 3E, the mRNA levels of four STAT3 downstream genes (Bcl-xL, Myc, Timp-1, and Cyclin D1) significantly decreased after the knockdown of EPHA5. A large number of studies have demonstrated that SRC and JAK2 are significant regulatory factors for STAT3 phosphorylation. To eliminate the possibility that SRC and JAK2 might be involved in the regulation of STAT3 by EPHA5, we examined the expression levels of SRC and JAK2 after EPHA5 knockdown. The results indicated that neither SRC nor JAK2 exhibited significant differences in expression (Supplementary Fig. 6).

A Interaction network with EPHA5, from Biogrid database. B Co-immunoprecipitation experiments were conducted on FTC-238 cells lysates with anti-EPHA5 and anti-STAT3, and then the precipitation was subsequently analyzed by western blotting with anti-STAT3 and anti-EPHA5. C Immunofluorescence analyses were performed using anti-EPHA5 antibodies or anti-STAT3 antibodies. Representative images are shown. Scale bar, 25 μm. D Western blot analysis of STAT3, p-STAT3, β-actin in FTC-133 and FTC-238 cells transfected with si-NC or si-EPHA5 respectively. **p < 0.01. n = 3. E QRT-PCR analysis of BCL-XL, Myc, Timp-1, and CyclinD1 mRNA in FTC-133 and FTC-238 cells with EPHA5 knockdown. *p < 0.05; **p < 0.01, n = 8.
Therefore, we concluded that EPHA5 is capable of binding to STAT3, activating STAT3, increasing levels of phosphorylated STAT3 (p-STAT3), and influencing the expression levels of genes related to the STAT3 pathway.
Knocking down EPHA5 has synergistic effect with SH-4-54
To further investigate the relationship between EPHA5’s role in promoting proliferation and inhibiting apoptosis of FTC cells and the STAT3 signaling pathway, we used the STAT3 phosphorylation (p-STAT3) inhibitor SH-4-54. Subsequently, we assessed changes in the expression of downstream proteins of STAT3 (STAT3, p-STAT3, PCNA). The results (shown in Fig. 4A) demonstrated that the expression of p-STAT3 and PCNA significantly decreased in the group treated with the p-STAT3 inhibitor (SH-4-54) alone, while the level of activated caspase-3 notably increased. As expected, cell cloning assay results indicated that SH-4-54 effectively inhibited the proliferative capacity of FTC cells (Fig. 4B). These findings suggested the effectiveness of the inhibitor. Combined treatment involving inhibiting EPHA5 expression with SH-4-54 resulted in a significant reduction in cell proliferation capacity and an increase in cell apoptosis compared to the group treated solely with SH-4-54 (Fig. 4A–C). Furthermore, similar conclusions can also be drawn from changes in cell morphology (Supplementary Fig. 2). To further clarify the role of the EPHA5/STAT3 axis, we repeated the above experiments by transfecting si-STAT3 and obtained experimental results similar to those with SH-4-54 (Fig. 4D–F).

A Western blot analysis of PCNA, p-STAT3, STAT3, Caspase-3, Cleaved-caspase3 and β-actin in FTC-133 and FTC-238 cells. *p < 0.05; **p < 0.01, n = 3. B Colony formation assay of FTC-133 and FTC-238 cell. **p < 0.01, n = 6. C Representative TUNEL staining of FTC cells; the nuclei were stained with DAPI. Scale bar = 200 μm. TUNEL scores. The relative number of apoptotic cells is represented as TUNEL-positive cells/DAPI. **p < 0.01, n = 6. D Western blot analysis of STAT3, cleaved-caspase-3, and β-actin in FTC-133 and FTC-238 cells. **p < 0.01, n = 3. E Colony formation assay of FTC-133 and FTC-238 cells transfected with si-NC or si-EPHA5 or si-EPHA5 + si-STAT3 respectively. *p < 0.05, **p < 0.01, n = 3. F Representative FITC and PI staining of FTC cells. Scale bar, 200 μm. The relative number of apoptotic cells is represented as FITC-positive cells ratio. **p < 0.01, n = 3.
These findings indicate that a synergistic effect between the inhibition of EPHA5 and SH-4-54 in suppressing cell proliferation and promoting apoptosis, implying that EPHA5 may have opposing effects compared to SH-4-54.
The function of EPHA5 in pro-proliferation and anti-apoptosis at least partly depend on the STAT3 signaling pathway in vivo and in vitro
To further investigate whether EPHA5’s role in proliferation and anti-apoptosis relies on the STAT3 signaling pathway. We conducted experiments by simultaneously overexpressing EPHA5 in FTC cells and adding SH-4-54. As expected, the expression levels of p-STAT3 and PCNA significantly decreased compared to the EPHA5-overexpression group (Fig. 5A). Subsequently, the colony assay results demonstrated that the promotion of proliferation due to the overexpression of EPHA5 was counteracted by SH-4-54 (Fig. 5B). Additionally, the results of the TUNEL assay indicated the same change (Fig. 5C). Subsequently, we replaced SH-4-54 with si-STAT3 for repeated experiments and obtained similar results (Fig. 5D–F). These findings suggest that the activation of STAT3 may be a prerequisite for EPHA5 to promote cell proliferation and inhibit cell apoptosis. To investigate whether similar phenomena occur in both in vivo and in vitro settings. We randomly assigned male nude mice into four groups, each comprising five mice. Administration of SH-4-54 significantly reduced tumor volume and weight (Fig. 5G–I). Overexpression of EPHA5 partially reversed the anti-cancer ability of SH-4-54. Additionally, the combination of SH-4-54 with EPHA5 knockdown demonstrated a stronger anti-cancer effect compared to the SH-4-54 alone group (Fig. 5G–I). Upon treatment with SH-4-54 after transfection with the EPHA5 overexpression plasmid, cell spread ability was compromised, and nuclear condensation occurred (Supplementary Fig. 2). Moreover, the apoptosis inhibitor Z-VAD attenuated the anti-proliferative capability of SH-4-54 against EPHA5 overexpression (Supplementary Fig. 3).

A Western blot analysis of p-STAT3, STAT3, PCNA, Caspase-3, Cleaved-caspase-3, β-actin in FTC-133 and FTC-238 cells. **p < 0.01, ##p < 0.01, n = 3. B Colony formation assay of FTC-133 and FTC-238 cells. **p < 0.01, ##p < 0.01, n = 6. C Representative TUNEL staining of FTC cells; the nuclei were stained with DAPI. Scale bar = 200 μm. TUNEL scores. The relative number of apoptotic cells is represented as TUNEL-positive cells/DAPI. *p < 0.05, ##p < 0.01, n = 6. D Western blot analysis of STAT3, cleaved-caspase-3, and β-actin in FTC-133 and FTC-238 cells. **p < 0.01, n = 3. E Colony formation assay of FTC-133 and FTC-238 cells transfected with si-NC or si-EPHA5 or si-EPHA5 + si-STAT3 respectively. **p < 0.01, n = 3. F Representative FITC and PI staining of FTC cells. Scale bar, 200 μm. The relative number of apoptotic cells is represented as FITC-positive cells ratio. *p < 0.05, n = 3. G Images of the xenograft tumors formed in nude mice subcutaneously injected with FTC-238 cells with EPHA5 knockdown or overexpression treated with SH-4-54. H, I Tumor weight was measured after the tumors were removed. Tumor growth curves were measured after injection of FTC238 cells with EPHA5 knockdown or EPHA5 overexpression treated with SH-4-54. *p < 0.05, ##p < 0.01, n = 5.
In summary, the function of EPHA5 in promoting proliferation and preventing apoptosis at least partly rely on the STAT3 signaling pathway both in vivo and in vitro (Fig. 6).

EPHA5 promotes cell proliferation and inhibits apoptosis in Follicular Thyroid Cancer via the STAT3 signaling pathway.
Discussion
According to 2016 data from the National Cancer Center, the incidence of thyroid cancer in China is steadily rising, consolidating its position as the most prevalent endocrine tumor with a rapidly increasing incidence [30, 31]. The main aim of this study was to elucidate novel pathogenic mechanisms underlying FTC and propose potential interventions. We provide groundbreaking evidence for two main findings: (1) Aberrant overexpression of EPHA5 in FTC. (2) The overexpression of EPHA5 substantially promoted FTC tumorigenesis both in vitro and in vivo, with these effects likely resulting from the activation of the STAT3 signaling pathway.
Studies have shown differential expression of EPHs family proteins in various tumors, such as liver cancer, prostate cancer, and ovarian cancer [32]. It has been proposed that EPHs family members can mediate intercellular signaling through their receptor function and binding to corresponding ligands in the membrane system [33, 34]. EPHs family members also have tyrosine kinase properties, although their complete spectrum of biological roles and underlying mechanisms has not been thoroughly investigated. EPHA5 has been implicated in the regulation of tumorigenesis in HER2-positive breast cancer and lung adenocarcinoma, and its association with mutations has been closely examined [35, 36]. Previous studies have emphasized the significance of EPHA5 in various human cancers, including lung cancer, prostate cancer, and ovarian cancer [37,38,39,40]. However, the expression levels and functions of EPHs in FTC are not clear. In this study, we detected the expression of EPHA5 in clinical FTC tissues and confirmed that its levels were higher compared to adjacent normal tissues. Furthermore, we discovered that EPHA5 serves as a predictor for poor prognosis in THCA, and its expression showed a positive correlation with the proliferation ability of FTC cells. After knocking down EPHA5, the proliferation of FTC-133/FTC-238 cells significantly decreased and apoptosis increased significantly, indicating that EPHA5 promotes FTC proliferation and inhibits apoptosis.
Aberrant activation of STAT3 is observed in more than 70% of human cancers [18, 19]. Previous studies have confirmed that STAT3, as a critical transcription factor, and the level of phosphorylation is linked to a poorer prognosis in various cancers, including prostate cancer, hepatocellular carcinoma, and pancreatic cancer [41,42,43]. The continuous activation of STAT3 results in a range of consequences, such as stimulating the proliferation of tumor cells, increasing their survival, preventing apoptosis, promoting metastasis, and facilitating tumorigenesis. Upstream signals (such as the cytokine family, activated STAT (PIAS) proteins, and protein tyrosine phosphatase (PTP) family) activate STAT3, leading to the formation of p-STAT3 dimers. These dimers then move into the nucleus where they regulate the transcription of target genes [44, 45]. The initial identification was made regarding the phosphorylation of the tyrosine residue (Y705) of STAT3. Subsequently, it was further determined that STAT3 phosphorylation on S694, S727, K170, and K685 also played significant roles in oncogenic functions [46]. Additionally, some studies have indicated the potential existence of various mechanisms underlying STAT3 activation in tumor cells [47, 48]. In our study, we found that the specific mechanism of action of EPHA5 was closely linked to the activation of STAT3. However, the precise mechanism underlying the interaction between EPHA5 and STAT3 remains unclear. Knocking down EPHA5 reduced the phosphorylation of STAT3, while overexpression of EPHA5 promoted the phosphorylation of STAT3. Furthermore, we observed that EPHA5, as one of the receptor tyrosine kinases (RTKs), had the ability to induce the phosphorylation of STAT3, subsequently increasing the expressions of downstream genes and promoting the proliferation of FTC cells. Simultaneously, the pro-proliferative and anti-apoptotic effects on FTC cells were significantly diminished after the combined the STAT3 inhibitor (SH-4-54)/si-STAT3, which interfered with the overexpression of EPHA5. On the other hand, knocking down EPHA5 in combination with SH-4-54/si-STAT3 further inhibited the proliferation of FTC cells and enhanced apoptosis. These results indicate that the pro-proliferative and anti-apoptotic effects of EPHA5 on FTC cells are closely linked to its mediated activation of STAT3. The oncogenic effect of EPHA5 relies, to a certain extent, on the activation of the STAT3 signaling pathway.
In our study, we have provided evidence suggesting that EPHA5 potentially regulates the tumor biology in FTC through promoting the phosphorylation modification of STAT3 in both in vivo and in vitro settings. Although the precise mechanism underlying the interaction between EPHA5 and STAT3 in FTC requires further experimental validation, our current discoveries offer a promising new target for genetic intervention in the treatment of FTC.
Limitations of the study
The specific phosphorylation site for activation of STAT3 by EPHA5 is not fully demonstrated in this paper. Further experimental evidence is needed to demonstrate whether the activation of STAT3 by EPHA5 is a direct effect or if there are some undisclosed mechanisms involved.
Data availability
The data supporting the findings of this study can be found in the article, or available from the corresponding author upon reasonable request.
References
Zheng RS, Zhang SW, Zeng HM, Wang SM, Sun KX, Chen R, et al. Cancer incidence and mortality in China, 2016. J Natl Cancer Cent. 2022;2:1–9.
Megwalu UC, Moon PK. Thyroid cancer incidence and mortality trends in the United States: 2000-2018. Thyroid J Am Thyroid Assoc. 2022;32:560–70.
Fallahi P, Ferrari SM, Elia G, Ragusa F, Patrizio A, Paparo SR, et al. Primary cell cultures for the personalized therapy in aggressive thyroid cancer of follicular origin. Semin Cancer Biol. 2022;79:203–16.
Denaro N, Nigro CL, Russi EG, Merlano MC. The role of chemotherapy and latest emerging target therapies in anaplastic thyroid cancer. OncoTargets Ther. 2013;9:1231–41.
Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717–20.
Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80.
Miao H, Wang B. EphA receptor signaling-complexity and emerging themes. Semin Cell Dev Biol. 2012;23:16–25.
Arvanitis DN, Davy A. Regulation and misregulation of Eph/ephrin expression. Cell Adhes Migr. 2012;6:131–7.
Nishimura M, Jung E-J, Shah MY, Lu C, Spizzo R, Shimizu M, et al. Therapeutic synergy between microRNA and siRNA in ovarian cancer treatment. Cancer Discov. 2013;3:1302–15.
Genander M, Halford MM, Xu N-J, Eriksson M, Yu Z, Qiu Z, et al. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell. 2009;139:679–92.
Huusko P, Ponciano-Jackson D, Wolf M, Kiefer JA, Azorsa DO, Tuzmen S, et al. Nonsense-mediated decay microarray analysis identifies mutations of EPHB2 in human prostate cancer. Nat Genet. 2004;36:979–83.
Cortina C, Palomo-Ponce S, Iglesias M, Fernández-Masip JL, Vivancos A, Whissell G, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat Genet. 2007;39:1376–83.
Batlle E, Bacani J, Begthel H, Jonkheer S, Gregorieff A, van de Born M, et al. EphB receptor activity suppresses colorectal cancer progression. Nature. 2005;435:1126–30.
Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, et al. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–28.
Fukuda A, Wang SC, Morris JP, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–55.
Ho PL, Lay EJ, Jian W, Parra D, Chan KS. Stat3 activation in urothelial stem cells leads to direct progression to invasive bladder cancer. Cancer Res. 2012;72:3135–42.
Kim DJ, Angel JM, Sano S, DiGiovanni J. Constitutive activation and targeted disruption of signal transducer and activator of transcription 3 (Stat3) in mouse epidermis reveal its critical role in UVB-induced skin carcinogenesis. Oncogene. 2009;28:950–60.
Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40:772–84.
Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007;251:199–210.
Roeser JC, Leach SD, McAllister F. Emerging strategies for cancer immunoprevention. Oncogene. 2015;34:6029–39.
Chen Y, Wang J, Wang X, Liu X, Li H, Lv Q, et al. STAT3, a poor survival predicator, is associated with lymph node metastasis from breast cancer. J Breast Cancer. 2013;16:40–9.
Kusaba T, Nakayama T, Yamazumi K, Yakata Y, Yoshizaki A, Inoue K, et al. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol Rep. 2006;15:1445–51.
Ludwig H, Nachbaur DM, Fritz E, Krainer M, Huber H. Interleukin-6 is a prognostic factor in multiple myeloma. Blood. 1991;77:2794–5.
Macha MA, Matta A, Kaur J, Chauhan SS, Thakar A, Shukla NK, et al. Prognostic significance of nuclear pSTAT3 in oral cancer. Head Neck. 2011;33:482–9.
Bromberg JF. Activation of STAT proteins and growth control. BioEssays. 2001;23:161–9.
Giordano TJ, Au AYM, Kuick R, Thomas DG, Rhodes DR, Wilhelm KG, et al. Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clin Cancer Res. 2006;12:1983–93.
Giordano TJ, Kuick R, Thomas DG, Misek DE, Vinco M, Sanders D, et al. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene. 2005;24:6646–56.
Zhang R, Liu J, Zhang W, Hua L, Qian L-T, Zhou S-B. EphA5 knockdown enhances the invasion and migration ability of esophageal squamous cell carcinoma via epithelial-mesenchymal transition through activating Wnt/β-catenin pathway. Cancer Cell Int. 2020;20:20.
Du J, Du Y, Chen L, Liu H. IL-17 promotes melanoma through TRAF2 as a scaffold protein recruiting PIAS2 and ELAVL1 to induce EPHA5. Biochim Biophys Acta Mol Cell Res. 2023;1870:119547.
Olson E, Wintheiser G, Wolfe KM, Droessler J, Silberstein PT. Epidemiology of thyroid cancer: a review of the National Cancer Database, 2000-2013. Cureus. 2019;11:e4127.
Nilubol N, Kebebew E. Should small papillary thyroid cancer be observed? A population-based study. Cancer. 2015;121:1017–24.
Khatami F, Larijani B, Nikfar S, Hasanzad M, Fendereski K, Tavangar SM. Personalized treatment options for thyroid cancer: current perspectives. Pharmacogenomics Pers Med. 2019;12:235–45.
Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. 2016;17:240–56.
Barquilla A, Pasquale EB. Eph receptors and ephrins: therapeutic opportunities. Annu Rev Pharm Toxicol. 2015;55:465–87.
Li Y, Chu J, Feng W, Yang M, Zhang Y, Zhang Y, et al. EPHA5 mediates trastuzumab resistance in HER2-positive breast cancers through regulating cancer stem cell-like properties. FASEB J. 2019;33:4851–65.
Huang W, Lin A, Luo P, Liu Y, Xu W, Zhu W, et al. EPHA5 mutation predicts the durable clinical benefit of immune checkpoint inhibitors in patients with lung adenocarcinoma. Cancer Gene Ther. 2021;28:864–74.
Wang H, Shan Q, Guo J, Han X, Zhao C, Li H, et al. PDL1 high expression without TP53, KEAP1 and EPHA5 mutations could better predict survival for patients with NSCLC receiving atezolizumab. Lung Cancer. 2021;151:76–83.
Li S, Zhu Y, Ma C, Qiu Z, Zhang X, Kang Z, et al. Downregulation of EphA5 by promoter methylation in human prostate cancer. BMC Cancer. 2015;15:18.
Staquicini FI, Qian MD, Salameh A, Dobroff AS, Edwards JK, Cimino DF, et al. Receptor tyrosine kinase EphA5 is a functional molecular target in human lung cancer. J Biol Chem. 2015;290:7345–59.
Chen X, Wang X, Wei X, Wang J. EphA5 protein, a potential marker for distinguishing histological grade and prognosis in ovarian serous carcinoma. J Ovarian Res. 2016;9:83.
Tai W-T, Chu P-Y, Shiau C-W, Chen Y-L, Li Y-S, Hung M-H, et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin Cancer Res. 2014;20:5768–76.
Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. Corrigendum. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Investig. 2015;125:1362.
Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017;394:76–87.
Masciocchi D, Gelain A, Villa S, Meneghetti F, Barlocco D. Signal transducer and activator of transcription 3 (STAT3): a promising target for anticancer therapy. Future Med Chem. 2011;3:567–97.
Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we?. Expert Opin Investig Drugs. 2009;18:45–56.
Debidda M, Wang L, Zang H, Poli V, Zheng Y. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J Biol Chem. 2005;280:17275–85.
Xu X, Han K, Zhu J, Mao H, Lin X, Zhang Z, et al. An inhibitor of cholesterol absorption displays anti-myeloma activity by targeting the JAK2-STAT3 signaling pathway. Oncotarget. 2016;7:75539–50.
Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6.
Funding
This study was supported by the Dalian Medical Science Research Plan Project (2112010) and “1 + X” project (2022 and 2022MDTQL02) of the Second hospital of Dalian Medical University.
Author information
Authors and Affiliations
Contributions
The primary experiments were carried out and data were analyzed by WGR and XGR. CJZ, XJC, CHJ, and LZF provided technical support. WGR, XGR, and CXW assisted in data analysis. The manuscript was written by WGR and XGR. The research was supervised and the manuscript was revised by WGZ, ZN, WGR and XGR. WGR, XGR and FYN contributed equally to this manuscript. All financial support was provided by WGZ and ZYF. The final manuscript was read and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All methods were performed in accordance with the relevant guidelines and regulations. All animal experiments were conducted under the approval of the Attitude of the Animal Care & Welfare Committee of Dalian Medical University (Approval number: ZN2021235).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, G., Xu, G., Fan, Y. et al. EPHA5 promotes cell proliferation and inhibits apoptosis in Follicular Thyroid Cancer via the STAT3 signaling pathway. Oncogenesis 14, 12 (2025). https://doi.org/10.1038/s41389-025-00556-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-025-00556-3
