
Abstract
While immunotherapy with immune checkpoint inhibitors (ICIs) has revolutionized the clinical management of various malignancies, a large fraction of patients are refractory to ICIs employed as standalone therapeutics, necessitating the development of combinatorial treatment strategies. Immunogenic cell death (ICD) inducers have attracted considerable interest as combinatorial partners for ICIs, at least in part owing to their ability to initiate a tumor-targeting adaptive immune response. However, compared with either approach alone, combinatorial regimens involving ICD inducers and ICIs have not always shown superior clinical activity. Here, we discuss accumulating evidence on the therapeutic interactions between ICD inducers and immunotherapy with ICIs in oncological settings, identify key factors that may explain discrepancies between preclinical and clinical findings, and propose strategies that address existing challenges to increase the efficacy of these combinations in patients with cancer.
Similar content being viewed by others

Introduction
Immune checkpoint inhibitors (Box 1) have revolutionized the clinical management of multiple cancer types, including (but not limited to) melanoma [1], non-small cell lung carcinoma (NSCLC) [2], and head and neck squamous cell carcinoma (HNSCC) [3]. However, even in oncological settings in which ICIs are approved by the Food and Drug Administration (FDA) and other regulatory agencies worldwide, a large fraction of patients are refractory to immunotherapy with ICIs [4]. Moreover, ICI administration can be associated with nonnegligible short- and long-term toxicities [5]. Thus, substantial efforts have been dedicated to the identification of effective and safe combinatorial partners for ICIs for a variety of cancer types [6].
In this context, considerable attention has been given to the possibility of combining ICIs with standard-of-care (SOC) therapeutic regimens encompassing conventional chemotherapy, radiation therapy (RT) and/or targeted anticancer drugs, largely reflecting (1) the established safety profile of these agents [7, 8], (2) their ability to promote (at least some degree) tumor debulking [9], and (3) at least in some settings, their capacity to mediate therapeutically relevant immunostimulatory effects [10,11,12]. Indeed, one of the major determinants of resistance to ICIs in patients with cancer is scarce infiltration of the tumor microenvironment (TME) at baseline by cytotoxic T lymphocytes (CTLs) [13], which often correlates with (1) a reduced tumor mutational burden and hence a low neoantigen load [14] and (2) limited expression of the coinhibitory ligand CD274 (best known as PD-L1) [15].
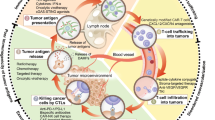
Several SOC therapeutics that can convert an immunologically “cold” (and hence ICI-insensitive) tumor into a “hot” neoplasm that exhibits an abundant CTL infiltrate and hence responds to ICIs (Fig. 1) belong to the class of immunogenic cell death (ICD, Box 2) inducers [16]. These therapeutics, which include specific chemical entities [10, 11, 17] as well as physical agents [18, 19], indeed, share the capacity to elicit a type of cell death that—in immunocompetent, syngeneic hosts—is sufficient to drive antigen-specific immune responses associated with an effector phase and the establishment of immunological memory [20].

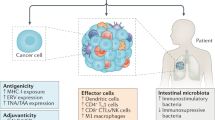
ICD as a tool for converting immunologically “cold” tumors into inflamed malignancies. Numerous biological, chemical and physical agents can elicit immunogenic cell death (ICD), a variant of regulated cell death that, in the context of failing to adapt to stress, antigenicity, adjuvanticity and permissive microenvironmental conditions, is sufficient to elicit adaptive immune responses specific for cell death-associated antigens that are associated with an active effector phase and the establishment of long-term immunological memory. Immunogenic cell death induction is generally associated with the abundant recruitment of immune effector cells and hence can (at least hypothetically) convert an immunologically cold tumor largely infiltrated by immunosuppressive M2-like tumor-associated macrophages (TAMs) and regulatory T (TREG) cells into an inflamed neoplasm exhibiting abundant infiltration by dendritic cells (DCs), cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. ANXA1 annexin A1, DAMP damage-associated molecular pattern, CALR calreticulin, CXCL10 C-X-C motif chemokine ligand 10, HMGB1, high mobility group box 1, IFN interferon
While some ICD inducers have been shown to positively cooperate with ICI-based immunotherapy in the clinic, others have largely failed to unlock the full therapeutic potential of ICIs in patients with cancer [16]. Here, we critically discuss preclinical and clinical evidence of successful or unsuccessful interactions between ICD-inducing agents and immunotherapy with ICIs in oncological settings, with a specific focus on potential strategies to ameliorate the clinical efficacy of these combinations.
Successful interaction between ICD inducers and immunotherapy with ICIs
In a number of preclinical and clinical settings, chemical and physical agents eliciting ICD have been shown to positively interact with ICIs, resulting in superior therapeutic effects compared with either approach alone. Interestingly, in multiple (especially preclinical) scenarios, superior efficacy has been obtained by administering ICD inducers and ICIs according to specific, context-dependent schedules [21] and by using ICD-inducing chemotherapeutics according to metronomic doses [22, 23].
Preclinical evidence
ICD-inducing conventional chemotherapeutics have been shown to cooperate with various ICIs in several syngeneic tumor models. For example, doxorubicin has been demonstrated to cooperate with ICIs specific for PD-L1, programmed cell death 1 (PDCD1, best known as PD-1), cytotoxic T lymphocyte-associated protein 4 (CTLA4) or CD96 in immunocompetent mice bearing subcutaneous 4T1 mammary carcinomas [24] or CT26 colorectal carcinomas (CRCs) [25, 26]. Similar results have been obtained by combining oxaliplatin with PD-1 or PD-L1 blockers in immunocompetent mice with subcutaneous MC38 or CT26 CRCs [26,27,28], PC3 prostate cancers [29], MOC1 HNSCCs [30], MB49 bladder tumors [31], H22 hepatocellular carcinomas [32], and MCA205 fibrosarcomas [33]. Notably, at least in some settings, such a therapeutic cooperativity could be further enhanced by immunostimulatory interventions beyond ICIs, including a Toll-like receptor 7 (TLR7) agonist [26, 34] and the systemic induction of autophagy with so-called caloric restriction mimetics (CRMs) [33, 35]. Similarly, a FOLFOX-mimicking chemotherapeutic regimen (which, among other components, includes oxaliplatin and 5-fluorouracil) reportedly synergizes with PD-1 blockers against CT26 CRCs developing in syngeneic immunocompetent BALB/c mice, a beneficial interaction that can be increased upon the codelivery of a monoclonal antibody specific for transforming growth factor beta 1 (TGFB1) [36].
Interestingly, while the ability of cisplatin to elicit bona fide ICD remains a subject of debate [37, 38], especially in clinical settings [39], this platinum derivative has been reported to synergize with a PD-1 blocker in immunocompetent mice bearing subcutaneous MOC1 HNSCCs [30], as well as with dual PD-1 and CTLA4 blockade in mice with established AB1 or AE17 mesotheliomas, resembling 5-fluorouracil [40]. The actual capacity of 5-fluororacil to cause ICD, however, is also controversial [41]. That said, both cisplatin and 5-fluorouracil have been shown to mediate therapeutically relevant immunostimulatory effects that may offer mechanistic ground for a positive interaction with ICIs [10]. Notably, another platinum derivative, PT-112, is a bone fide ICD inducer and indeed has been shown to synergize with PD-1 as well as PD-L1 blockers against subcutaneous TS/A mammary carcinomas developing in syngeneic immunocompetent BALB/c mice [42], suggesting that the ICD-inducing potential of platinum compounds depends on specific molecular features of the coordination complex and its cellular effects rather than on the platinum ion itself [10].
Other ICD-inducing chemotherapeutics that have been shown to synergize with ICIs in preclinical tumor models include (but are not limited to): (1) cyclophosphamide, which reportedly cooperates with PD-1 or CTLA4 blockers, optionally in the context of extra immunotherapeutic strategies such as adoptive cell transfer and vaccination, in mice bearing subcutaneous A20HA lymphomas [43] TC-1 lung carcinomas [44] or CT26 CRCs [45]; (2) mitoxantrone, which has been demonstrated to engage in positive therapeutic interaction with a PD-1 blockers in MCA205 fibrosarcoma-bearing mice, especially when combined with CRMs [33]; as well as (3) paclitaxel, which has been reported to cooperate with a PD-1 blocker against subcutaneous E0771 mammary carcinomas established in C57BL/6 mice [46].
For physical ICD inducers [47], RT delivered according to a hypofractionated schedule to a single neoplastic lesion has been demonstrated to synergize with CTLA4 and/or PD-1 blockers in immunocompetent BALB/c mice bearing bilateral TS/A mammary carcinomas or 344SQ lung carcinomas s.c., ultimately resulting in (at least some degree of) control of the contralateral, nonirradiated lesion [48,49,50,51], as well as in wild-type BALB/c mice with subcutaneous 4T1 mammary carcinomas, culminating with partial control of metastatic lung nodules [52]. In the 4T1 model, similar results have been obtained by combining RT with an ICI specific for V-set immunoregulatory receptor (VSIR, best known as VISTA) and metronomic cyclophosphamide [53]. Moreover, superior therapeutic benefits have been documented when RT was delivered in the context of CTLA4 or PD-1 blockade along with the following: (1) cyclophosphamide or doxorubicin, in immunocompetent C57BL/6 mice with subcutaneous mEER HNSCCs, B16 melanomas or MC38 CRCs [54, 55]; (2) the oncolytic peptide LTX-315 [56], in wild-type BALB/c mice bearing bilateral subcutaneous TS/A mammary carcinomas [57]; and (3) an agonistic antibody targeting TNF receptor superfamily member 9 (TNFRSF9, best known as CD137 or 4-1BB) or CD40 plus cyclophosphamide, in immunocompetent mice bearing AT3 mammary tumors s.c. or orthotopic ID8 ovarian carcinomas, respectively [58, 59]. Similarly, while photodynamic therapy has been shown to positively interact with PD-1 or PD-L1 blockers in various syngeneic mouse models of mammary carcinoma [60, 61] and CRC [60], low-dose pulsed ultrasound has been reported to synergize with a monoclonal antibody specific for PD-1 and doxorubicin in immunocompetent mice bearing orthotopic CT2A or GL261 glioblastomas [62].
Importantly, data from preclinical tumor models suggest that ICD inducers, as well as other (immuno)therapeutics, engage in superior interactions with ICIs when (1) the former are administered at low doses or according to metronomic schedules, rather than in an attempt to achieve a maximum-tolerated dose (MTD) [27, 32, 40], and/or (2) the former are administered according to specific schedules with respect to the latter, in a highly context-dependent manner [27, 31, 32, 63]. For example, mice bearing MC38 CRCs s.c. have been reported to respond to ICI-based immunotherapy only once previously administered a low dose (10 mg/kg) but not an ultralow (5 mg/kg) or high (20 mg/kg) dose of oxaliplatin, culminating in improved tumor infiltration by CD8+ CTLs, superior secretion of interferon gamma (IFNG), and consequently long-term disease control associated with the development of immunological memory [27]. Similarly, decreasing the total RT dose or RT dose per fraction has been associated with superior immunogenicity and hence synergistic interactions with ICIs in a variety of preclinical tumor models, including immunocompetent mice bearing TS/A mammary carcinomas [48, 50], ID8 ovarian carcinomas [59], and 344SQ lung adenocarcinomas [51]. Moreover, a single administration of low-dose oxaliplatin (3 mg/kg) has been shown to sensitize C57BL/6 mice bearing MC38 CRCs to ICIs targeting CTLA4 or PD-1 when delivered concurrently [64]. Similar results have been obtained with the concurrent (but not sequential) delivery of ICD-inducing chemotherapeutics or fractionated RT and an ICI targeting PD-L1 in preclinical models of CRC (CT26), triple-negative breast cancer (TNBC) (4T1) and glioblastoma [26, 65]. Conversely, the sequential (but not concurrent) administration of oxaliplatin upfront followed by a PD-1 blocker has been associated with superior therapeutic interactions in mice bearing MC38 CRCs [27]. Moreover, the delivery of cyclophosphamide before an ICI specific for CTLA4 has been demonstrated to eradicate CT26 CRCs established s.c. in immunocompetent BALB/c mice, an effect that was lost by the swapping administration schedule [45].
In summary, abundant preclinical data support the notion that ICD inducers can positively cooperate with ICIs in vivo, with a major, context-dependent impact for dose and administration schedule.
Clinical evidence
A number of combinatorial therapeutic regimens involving one or more ICD inducer(s) (generally administered according to standard MTD approaches) and an ICI are currently approved by the FDA and other regulatory agencies worldwide for use in patients with cancer [66, 67], strongly supporting the notion that (at least in some oncological indications) the ability of cancer cells undergoing ICD to inflame the TME promotes ICI sensitivity (Table 1).
The IMpassion130 trial, in which patients with metastatic TNBC were randomly allocated to nab-paclitaxel plus placebo or atezolizumab (a PD-L1 blocker), demonstrated a significant overall survival (OS) advantage for individuals bearing PD-L1+ tumors in the combination arm (median OS: 25.0 vs. 15.5 months), an effect that was reduced in the intention-to-treat population [68]. Although exploratory, these data were largely confirmed in the ANASTASE phase III study [69], paving the way for the approval of this regimen as a first-line therapy for patients with PD-L1+ TNBC, even though the IMpassion131 trial failed to recapitulate the findings of IMpassion130 [70], which led the FDA to issue an official alert (https://www.fda.gov/drugs/resources-information-approved-drugs/fda-issues-alert-about-efficacy-and-potential-safety-concerns-atezolizumab-combination-paclitaxel#:~:text=On%20September%208%2C%202020%2C%20the,or%20metastatic%20triple%20negative%20breast). Multiple randomized, phase III clinical trials reported a progression-free survival (PFS) and/or an OS survival advantage for patients with TNBC receiving SOC chemotherapy plus pembrolizumab vs chemotherapy alone. For example, the KEYNOTE-355 trial documented both a PFS and an OS advantage in the PD-L1+ patient population (PFS: 9.7 months vs 5.6 months; OS: 23.0 months vs 16.1 months) [71], a beneficial effect that was less pronounced in the intention-to-treat population [71] and largely confirmed by the CheckMate 648 [72] and KEYNOTE-859 [73] trials. Similarly, the PD-1 blocker nivolumab combined with FOLFOX (folinic acid, 5-fluorouracil, and oxaliplatin) or XELOX (capecitabine and oxaliplatin) has been shown to provide superior PFS and OS advantages to previously untreated patients with HER2+ advanced gastric cancer, gastroesophageal junction cancer and esophageal adenocarcinoma enrolled in the CheckMate 649 trial, especially in subjects with a PD-L1 combined positive score ≥ 5, obtaining regulatory approval as first-line therapy for these oncological indications [74].
Alongside concurrent treatment schedules, therapeutic regimens combining an ICD inducer upfront followed by an ICI have also been shown to provide superior clinical benefits in various oncological settings. Thus, the phase II TONIC trial aimed to determine the best ICD inducer to condition the TME of patients with TNBC to maximize the clinical response to nivolumab [75]. Specifically, these patients were randomly allocated to receive hypofractionated RT in 3 fractions of 8 Gy each, cyclophosphamide (50 mg orally daily for 2 weeks), cisplatin (40 mg/m2 weekly for 2 weeks), or doxorubicin (15 mg weekly for 2 weeks), followed by SOC nivolumab administration. Across treatment arms, the overall response rate (ORR) was 20%, and even though cyclophosphamide and cisplatin followed by nivolumab were associated with a lower ORR than nivolumab monotherapy was, low-dose doxorubicin appeared to effectively sensitize TNBC to immune checkpoint inhibition (ORR: 35%), comparing well to nivolumab monotherapy (ORR: 17%) [75]. Similarly, the phase III PACIFIC trial documented a significant PFS (16.8 months vs 5.6 months) and OS (3-year OS: 57% vs 43.5%) advantage in patients with unresectable stage III NSCLC who did not progress on definitive, platinum-based, chemoradiation therapy subsequently receiving durvalumab (a PD-L1 blocker) vs placebo [76, 77]. While whether such an effect depends on a positive therapeutic interaction between chemoradiation and durvalumab rather than on the activity of the latter remains to be determined, these findings led to the approval of durvalumab as an SOC for patients with unresectable stage III NSCLC following chemoradiotherapy, marking a significant advancement in the treatment of this challenging patient population.
Recent clinical findings also support the use of neoadjuvant chemotherapy-immunotherapy combinations, especially durvalumab-based regimens, in patients with TNBC [78, 79]. Patients enrolled in the phase II GeparNuevo trial, which were randomly allocated to receive a single durvalumab injection prior to or along with chemotherapy with nab-paclitaxel, epirubicin, or cyclophosphamide followed by surgery, did not achieve a pathological complete response more frequently than patients receiving neoadjuvant chemotherapy only (53% vs 44%, not significant) [80]. However, compared with patients treated with neoadjuvant chemotherapy alone, individuals in the study arm that included durvalumab had significant benefits in terms of 3-year invasive disease-free survival (85.6% vs 77.2%), 3-year distant disease-free survival (91.7% vs 78.4%) and 3-year OS (95.2% vs 83.5%) [78, 80]. Interestingly, tumor-infiltrating lymphocytes were increased by treatment in both study arms [80], suggesting that while blocking PD-1 may activate a clinically relevant TNBC-targeting immune response in this patient population, ICD induction by chemotherapy may be required to enable abundant tumor infiltration by immune effector cells. Similarly, the open-label phase III CheckMate 816 trial, which compared neoadjuvant chemotherapy (either carboplatin plus paclitaxel, gemcitabine plus cisplatin, or pemetrexed plus cisplatin) combined with nivolumab to neoadjuvant chemotherapy only in patients with NSCLC, documented statistically significant benefits for ICI-containing regimens in terms of median event-free survival (31.6 months vs 20.8 months) and pathological complete response rates (24.0% vs 2.2%) [81]. Similar results have been obtained in the Phase III KEYNOTE-671 [82] and NADIMII [83] trials. Moreover, impressive results have also been achieved with neoadjuvant ICI-based immunotherapy alone in patients with melanoma [84,85,86], locally advanced HNSCC [87], and locally advanced CRC [88, 89], potentially suggesting that, at least in some patient populations, neoadjuvant ICIs may not require ICD induction to enable major clinical responses, a possibility that remains to be investigated in the TNBC setting.
In summary, combinatorial regimens involving one or more ICD inducer(s) and ICI-based immunotherapy appear to be superior to ICD inducers alone in a variety of clinical scenarios, but whether this truly reflects a positive therapeutic interaction rather than a pronounced efficacy of (at least some) ICIs employed as monotherapy generally remains to be formally investigated in patients.
Unsuccessful interaction between ICD inducers and immunotherapy with ICIs
Not all preclinical and clinical studies completed thus far have documented a successful interaction between ICD induction by conventional chemotherapeutics, RT or targeted anticancer agents and ICI-based immunotherapy, calling for an improved understanding of the underlying (immuno)biological reasons for the design of optimized combinatorial regimens for clinical use.
Preclinical evidence
Even in preclinical tumor models, ICD inducers and ICIs do not necessarily exhibit cooperative effects, at least in some cases owing to inappropriate dosing or administration schedules that promote the intratumoral or systemic accumulation of immunosuppressive cells.
In contrast to E0771 mammary carcinomas [46], MC38 CRCs established in immunocompetent C57BL/6 mice do not exhibit superior responses to the simultaneous administration of paclitaxel (20 mg/kg) and docetaxel (10 mg/kg) in combination with a PD-1 blocker, a lack of therapeutic interaction that appears to be accompanied by scarce recruitment of tumor-infiltrating lymphocytes to the tumor bed [90]. The concurrent administration of oxaliplatin (~10 mg/kg) plus a PD-L1 blocker to mice bearing subcutaneous CT26 CRCs also failed to significantly affect tumor growth and did not improve the intratumoral or systemic ratio between CD8+ T cells and immunosuppressive CD4+CD25+FOXP3+ regulatory T (TREG) cells [28, 91], indicating that additional immunostimulatory molecules, such as TLR7 agonists, may be required to enable this therapeutic interaction [26]. Moreover, the efficacy of both carboplatin (100 mg/kg) and paclitaxel (10 mg/kg) against mouse ID8 ovarian cancers established intraperitoneally has been shown to remain unaltered by the concomitant administration of an ICI targeting hepatitis A virus cellular receptor 2 (HAVCR2, also known as TIM-3) [92]. At least hypothetically, the inability of taxanes and oxaliplatin to cooperate with ICIs in immunocompetent mice bearing CRCs or ovarian cancers may reflect tumor-intrinsic features that prevent the optimal induction of ICD in vivo. This possibility, however, remains to be formally investigated.
Interestingly, in the latter experimental setting, employing a sequential (rather than concurrent) delivery schedule resulted in inferior efficacy coupled with the intraperitoneal accumulation of TREG cells [92]. Similarly, while delivering cyclophosphamide (100 mg/kg) upfront reportedly synergizes with a CTLA4 blocker in CT26-bearing mice, inverting the order of administration appears to compromise therapeutic interactions along with an increase in CD8+ T-cell apoptosis [45, 93]. Notably, the same combinatorial regimen does not exhibit any efficacy in mice bearing subcutaneous RENCA renal cell carcinomas, which is at least correlated with the accumulation of myeloid-derived suppressor cells (MDSCs) and the consequent release of immunosuppressive cytokines [45, 94].
At least in some preclinical tumor models, including subcutaneous MC38 CRCs and H22 hepatocellular carcinomas growing in immunocompetent hosts, cisplatin also appears unable to unlock full-blown therapeutic responses to PD-1 blockers, irrespective of relative administration schedule [27, 32], which may reflect the limited ability of cisplatin to elicit ICD in some settings [37]. Comparable results have been obtained with in vivo syngeneic mesothelioma models treated with vinorelbine or 5-fluorouracil and dual PD-1/CTLA4 blockade, with AB1 (but not AE17) mesothelioma revealing some degree of antagonism [40]. Similarly, paclitaxel delivered intraperitoneally at the MTD (50 mg/kg) reportedly fails to cooperate with a PD-L1 blocker in immunocompetent C57BL/6 mice bearing EO771 TNBCs [95]. While paclitaxel has been shown to unlock the therapeutic efficacy of a PD-1 blocker in the same model [46], whether this apparent discrepancy reflects intrinsic immunobiological differences between PD-L1 and PD-1 signaling remains to be fully elucidated.
Collectively, these preclinical studies highlight the complexity of optimizing combinatorial regimens involving one or more ICD inducer(s) and ICIs, as an inappropriate dose or administration schedule can not only prevent these agents from cooperating but also (at least in some settings) can generate therapeutic antagonism, often in the context of local or systemic immunosuppression.
Clinical evidence
Despite considerable expectations, numerous randomized phase II or III clinical trials have failed to demonstrate an advantage from combining ICIs with SOC ICD-inducing therapeutic regimens (Table 2).
The IMpower131 and IMpower132 trials explored the addition of atezolizumab to SOC chemotherapeutic regimens with ICD-inducing activity in patients with NSCLC, highlighting potential benefits but also inconsistent improvements in disease outcome [96,97,98]. IMpower131 enrolled patients with advanced squamous NSCLC and compared the efficacy of atezolizumab vs placebo combined with carboplatin and either paclitaxel or nab-paclitaxel. While PFS was significantly improved in the atezolizumab arm, particularly in patients with PD-L1+ lesions, there was no significant extension in OS [96]. Virtually identical findings emerged from IMpower132, which randomly allocated patients with advanced nonsquamous NSCLC to atezolizumab vs placebo plus platinum-based chemotherapy (carboplatin or cisplatin plus pemetrexed) [97]. Moreover, the addition of the CTLA4 blocker ipilimumab to paclitaxel- and carboplatin-based chemotherapy failed to improve disease outcomes in two randomized, phase III clinical trials enrolling patients with squamous NSCLC (NCT01285609, NCT02279732) while significantly increasing the incidence of adverse events [99]. The large-scale CA184-156 trial tested the combination of etoposide and platinum-based chemotherapy optionally in combination with ipilimumab (followed by ipilimumab maintenance) in patients with extensive small lung cell carcinoma (SCLC), with outcomes that did not differ across study arms [100]. Similar findings were documented by the CheckMate 451 and CheckMate 331 studies, two randomized, phase III trials that enrolled patients with SCLC receiving first-line platinum-based chemotherapy (not earlier than 3 weeks later) with nivolumab, ipilimumab or placebo (CheckMate 451) [101], or with SCLC relapsing after platinum-based chemotherapy treated with nivolumab, topotecan, or amrubicin (CheckMate 331) [102]. Indeed, despite the lack of a placebo arm in the latter study, ICI-based immunotherapy delivered after ICD-inducing chemotherapy failed to enable clinical advantages, potentially owing to the delay between the two approaches.
Considerable challenges have also been documented in gastroesophageal and colorectal settings. For example, while the phase III KEYNOTE-062 trial, which investigated the combination of pembrolizumab plus chemotherapy (cisplatin plus 5-fluorouracil or capecitabine) in previously untreated patients with PD-L1+ advanced gastric or gastroesophageal junction adenocarcinoma, demonstrated a higher ORR in patients allocated to the combination treatment arm, it failed to result in improved PFS, OS, or response duration (but increased the rate and severity of side effects) [103, 104]. Moreover, the randomized phase II/III CheckMate 9×8 trial, which explored the addition of nivolumab to SOC FOLFOX plus bevacizumab-based chemotherapy in previously untreated patients with unresectable CRC, demonstrated a trend toward an improved 1-year PFS rate, ORR and response duration for the combinatorial regimen over SOC chemotherapy only but failed to meet its primary endpoint of PFS extension [105, 106]. Nivolumab not only failed to ameliorate disease outcomes in patients with newly diagnosed glioblastoma with a methylated or undetermined MGMT promoter subjected to surgery plus adjuvant RT plus temozolomide-based chemotherapy in the context of a randomized phase III CheckMate 548 clinical trial [107] but also failed to outperform temozolomide as a partner for adjuvant RT in patients with newly diagnosed glioblastoma with an unmethylated MGMT promoter in the context of the randomized phase III CheckMate 498 study [108]. Finally, the phase III KEYNOTE-921 trial demonstrated that the therapeutic activity of docetaxel in patients with metastatic castration-resistant prostate cancer (CRPC) cannot be ameliorated by the coadministration of pembrolizumab or ipilimumab [109], as did the randomized CA184-043 study, which randomly allocated men with metastatic CRPC who failed docetaxel-based chemotherapy and received bone-targeting RT to ipilimumab or placebo [110].
Trials investigating ICD-chemotherapeutics and ICIs in patients with urothelial carcinoma have faced similar obstacles. Neither pembrolizumab nor atezolizumab were able to improve OS extension afforded by carboplatin- or cisplatin-based SOC chemotherapy (in one study, optionally combined with gemcitabine) in patients with urothelial carcinoma enrolled in the randomized, phase III KEYNOTE-361 [111] and IMvigor130 [112] clinical studies, despite at least some PFS benefit associated with the administration of atezolizumab plus gemcitabine and platinum-based (especially cisplatin-based) chemotherapy [112]. Similarly, two randomized clinical trials investigating avelumab as a therapeutic partner for SOC chemoradiation in patients with locally advanced HNSCC (JAVELIN 100 and GORTEC-REACH) failed to document an OR benefit for the combinatorial regimens over SOC chemoradiation alone [113, 114]. Moreover, IMpassion131, a randomized phase III clinical study testing paclitaxel with atezolizumab or placebo in patients with TNBC, failed to demonstrate any improvement in PFS or OS for the combinatorial regimen over SOC chemotherapy, although these patients also received dexamethasone, which is a powerful immunosuppressant often required to limit adverse events (be they elicited by natural disease progression or treatment) in patients [115, 116], before at least the first two infusions of paclitaxel [70]. Similarly, neoadjuvant carboplatin plus nab-paclitaxel and atezolizumab, followed by an adjuvant anthracycline regimen, was not more efficient than the same regimen without atezolizumab and did not ameliorate pathological complete response among TNBC patients enrolled in the NeoTRIP Michelangelo randomized trial [117]. Finally, patients with high-risk, early HER2+ breast cancer receiving neoadjuvant atezolizumab in combination with dose-dense doxorubicin plus cyclophosphamide, followed by paclitaxel, trastuzumab, and pertuzumab enrolled in the Phase III IMpassion050 trial failed to experience pathological complete responses at increased rates compared with similarly treated women who received placebo instead of atezolizumab, neither in the intention-to-treat population nor among subjects with PD-L1+ tumors, ultimately leading to study discontinuation because of an unfavorable risk‒benefit ratio [118]. Similar dismal findings have been obtained by the randomized phase III IMagyn050 trial, which compared the efficacy of neoadjuvant atezolizumab vs placebo plus paclitaxel, carboplatin, and bevacizumab, followed by adjuvant bevacizumab, in patients with stage III/IV ovarian carcinoma [119].
Collectively, these observations suggest that ICD-inducing chemotherapeutic and radiotherapeutic regimens do not always cooperate with ICIs in the clinic, highlighting a critical need to understand the obstacles that currently limit the translation of existing preclinical data toward the development of combinatorial regimens with superior efficacy in patients, at least partially through changes in dose and administration schedule.
Concluding remarks
In summary, an expanding preclinical literature suggests that various ICD-inducing cancer therapeutics can positively interact with ICIs across a panel of malignancies (Fig. 2). That said, such a positive interaction may not necessarily emerge from ICD induction but may rather reflect ICD-unrelated immunostimulatory effects that may support ICI sensitivity, as in vivo ICD induction remains challenging to assess [11, 120]. Moreover, despite such abundant preclinical findings, ICIs ameliorated the clinical efficacy of ICD-inducing anticancer agents delivered according to SOC dose and administration schedules in only a few clinical scenarios.

Potential synergy between ICD inducers and ICIs. Owing to their ability to recruit immune effector cells, including dendritic cells (DCs) and cytotoxic T lymphocytes (CTLs), immunogenic cell death (ICD)-inducing regimens (at least in some settings) can convert immunologically cold tumors into inflamed lesions. In this context, immune checkpoint inhibitors (ICIs), such as monoclonal antibodies targeting programmed cell death 1 (PDCD1, best known as PD-1) or CD274 (best known as PD-L1), which normally operate by (re)activating CTLs, may exhibit superior efficacy, providing a solid rationale for developing combinatorial clinical strategies potentially associated with improved clinical outcomes; CALR calreticulin, IFN interferon
Several factors may explain (at least in part) such a discrepancy between preclinical and clinical settings (Fig. 3). First, while in preclinical settings, ICD-inducing therapeutics can be easily delivered according to nonconventional doses, including metronomic schedules that have been consistently associated with increased immunogenicity [121,122,123,124], clinical trials combining ICD inducers and ICIs generally rely on SOC approaches, which have often been developed according to the MTD principle and hence may be associated with nonnegligible lympho- and myelosuppression [125,126,127,128,129]. In this context, it would be fundamental to test ICIs plus ICD inducers administered according to metronomic schedules or at doses lower than the MTD in clinical settings that may be compatible with such an approach, for example, in patients experiencing severe adverse events when ICD-inducing agents are delivered as per SOC. Second, most preclinical studies testing ICD inducers plus ICIs harness mouse cancer cell lines to establish subcutaneous tumors in syngeneic immunocompetent hosts, which (1) largely fail to recapitulate the intra- and interpatient heterogeneity of human tumors [130], and (2) offer to dying cancer cells a privileged and most often nonphysiological immunological contexture to elicit anticancer immunity [131, 132]. The use of mouse tumor models that develop orthotopically in the context of failing immunosurveillance, such as carcinogen-elicited or genetically driven neoplasms, may circumvent (at least in part) these limitations [133,134,135].

Advantages and limitations of current mouse models for the study of ICD. Most mouse models currently employed to investigate immunogenic cell death (ICD) induction are amenable to testing multiple (including noncanonical) dose regimens and administration schedules, offer rapid turnaround times and are compatible with the formal assessment of tumor-targeting immune responses. However, these models do not necessarily recapitulate human oncogenesis in terms of disease site or inter- and intratumor heterogeneity. Moreover, whether the murine system can properly model ICD induction in human cancer cells as well as whether systemic factors may influence immune fitness in patients with cancer remains to be formally established
Third, most often, clinical trial design fails to build on preclinical data comparing different administration schedules for combining ICD inducers with ICIs (e.g., concurrent vs. sequential with ICIs first-in vs. sequential with ICD induction first-in) to achieve superior efficacy, which tends to exhibit at least some context dependency [21, 136]. A systematic preclinical assessment of administration schedules in immunocompetent tumor models is expected to assist in the identification of optimal regimens to combine ICD inducers with ICIs for translation to clinical testing, potentially reducing the number of trials ultimately reporting a lack of interaction between these treatment modalities. Fourth, ICD induction by chemotherapy, RT or targeted anticancer agents as formally assessable only in syngeneic mouse tumor models [137] may not necessarily result in similar efficacy in fully human systems (and notably cancer patients), potentially calling for the development of combinatorial ICD-inducing strategies. As a standalone example, cisplatin is a poor ICD inducer but may be converted into a powerful inducer by combining it with an endoplasmic reticulum stressor [37]. Fifth, while ICD induction in vitro is fairly straightforward, human tumors evolve as they establish numerous, not necessarily overlapping, mechanisms that limit the induction of ICD and its perception as immunogenic by the host [138,139,140]. Identifying these mechanisms, which may vary not only across tumor types but also across different malignant lesions in the same patient or even across different areas of the same tumor, on an individual basis may offer actionable mechanistic insights to develop superior combinations of ICD inducers and ICIs. Sixth, a number of variables affecting patient immune fitness may prevent ICD inducers from actually eliciting an ICI-active immune response, including (1) polymorphisms in genes encoding critical immune receptors [141], (2) alterations in the gut or intratumoral microbiome [142], (3) dietary habits [143], (4) comorbidities [144], and (5) medications and over-the-counter drugs [115, 145, 146]. Upon precise identification, many of these barriers may offer a means to (1) select patients at increased likelihood to benefit from therapeutic regimens involving ICD-inducing agents and ICIs and/or (2) improve the efficacy of such combinatorial strategies.
Finally, cancer cells exhibit extraordinary heterogeneity, not only across tumor types or in different patients with the same neoplasms but also across different tumors in the same patient and even within individual lesions [130]. This implies that specific therapeutics may elicit ICD in some but not all cancer cells, at least in part reflecting the high interconnectivity that characterizes cell death signaling modules, which ultimately impacts immunogenicity [147,148,149]. While spatially resolved omics technologies may offer an improved characterization of the heterogeneity of malignant lesions with respect to transcriptional, proteomic and metabolomic features [150], whether any of these parameters or combinations thereof may accurately predict the propensity of an individual tumor to respond to ICD inducers alone or combined with ICIs has yet to be demonstrated. Similarly, while a number of circulating factors are being scrutinized for their prognostic and predictive value in different oncological indications [151], whether these biomarkers can be used to efficiently identify patients with cancer at an increased likelihood of benefiting from ICD-inducing therapeutics in combination with ICIs remains unclear. As an added layer of complexity, a surge in the circulating levels of ICD-associated biomarkers such as high mobility group box 1 (HMGB1), which has been correlated with improved disease outcome in patients with breast carcinoma or HNSCC receiving ICD-inducing agents [152, 153], may de facto originate from ICD-unrelated processes, hence potentially being poorly predictive of a positive interaction with ICIs.
As such, outstanding challenges for the field include (but are not limited to): (1) the identification of new chemical entities or physical agents with superior ICD-inducing capacity that can be moved forward to clinical translation; (2) the characterization of novel, clinically relevant dosing schedules to increase the ICD-inducing potential of anticancer therapeutics commonly used in the clinic at (or close to) the MTD; (3) the deconvolution of novel cellular pathways leading to bona fide ICD; (4) the identification of preclinical tumor models that recapitulate the cancer‒immunity interaction as closely as possible to their human counterparts; (5) the use of such models toward an unbiased assessment of optimal combinatorial regimens with respect to the administration schedule; (6) the development of strategies that circumvent the natural tendency of human tumors to evade immunosurveillance; and (7) the identification of host of cancer-related factors that limit the perception of cell death as immunogenic and at the same time may be amenable to therapeutic targeting or aid patient stratification.
In conclusion, while additional preclinical and clinical work is needed to unlock the full therapeutic potential of ICD-inducing therapeutics as partners for ICIs, we surmise that addressing these obstacles, or at least taking them under attentive consideration as potential predictors of response during clinical trial design, may lead to the development of novel, safe and efficient combinatorial regimens for patients with a wide variety of malignancies.
References
Huang AC, Zappasodi R. A decade of checkpoint blockade immunotherapy in melanoma: understanding the molecular basis for immune sensitivity and resistance. Nat Immunol. 2022;23:660–70. https://doi.org/10.1038/s41590-022-01141-1.
Mountzios G, Remon J, Hendriks LEL, García-Campelo R, Rolfo C, Van Schil P, et al. Immune-checkpoint inhibition for resectable non-small-cell lung cancer - opportunities and challenges. Nat Rev Clin Oncol. 2023;20:664–77. https://doi.org/10.1038/s41571-023-00794-7.
Cohen EEW, Bell RB, Bifulco CB, Burtness B, Gillison ML, Harrington KJ, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J Immunother Cancer. 2019;7:184. https://doi.org/10.1186/s40425-019-0662-5.
Mao Y, Xie H, Lv M, Yang Q, Shuang Z, Gao F, et al. The landscape of objective response rate of anti-PD-1/L1 monotherapy across 31 types of cancer: a system review and novel biomarker investigating. Cancer Immunol Immunother. 2023;72:2483–98. https://doi.org/10.1007/s00262-023-03441-3.
Sullivan RJ, Weber JS. Immune-related toxicities of checkpoint inhibitors: mechanisms and mitigation strategies. Nat Rev Drug Discov. 2022;21:495–508. https://doi.org/10.1038/s41573-021-00259-5.
Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18:197–218. https://doi.org/10.1038/s41573-018-0007-y.
De Ruysscher D, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F. Radiotherapy toxicity. Nat Rev Dis Primers. 2019;5:13. https://doi.org/10.1038/s41572-019-0064-5.
Kuderer NM, Desai A, Lustberg MB, Lyman GH. Mitigating acute chemotherapy-associated adverse events in patients with cancer. Nat Rev Clin Oncol. 2022;19:681–97. https://doi.org/10.1038/s41571-022-00685-3.
Dall'Olio FG, Marabelle A, Caramella C, Garcia C, Aldea M, Chaput N, et al. Tumour burden and efficacy of immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2022;19:75–90. https://doi.org/10.1038/s41571-021-00564-3.
Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725–41. https://doi.org/10.1038/s41571-020-0413-z.
Petroni G, Buqué A, Coussens LM, Galluzzi L. Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat Rev Drug Discov. 2022;21:440–62. https://doi.org/10.1038/s41573-022-00415-5.
Petroni G, Cantley LC, Santambrogio L, Formenti SC, Galluzzi L. Radiotherapy as a tool to elicit clinically actionable signalling pathways in cancer. Nat Rev Clin Oncol. 2022;19:114–31. https://doi.org/10.1038/s41571-021-00579-w.
Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80. https://doi.org/10.1038/s41568-020-0285-7.
Budczies J, Kazdal D, Menzel M, Beck S, Kluck K, Altbürger C, et al. Tumour mutational burden: clinical utility, challenges and emerging improvements. Nat Rev Clin Oncol. 2024;21:725–42. https://doi.org/10.1038/s41571-024-00932-9.
Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18:345–62. https://doi.org/10.1038/s41571-021-00473-5.
Galluzzi L, Guilbaud E, Schmidt D, Kroemer G, Marincola FM. Targeting immunogenic cell stress and death for cancer therapy. Nat Rev Drug Discov. 2024;23:445–60. https://doi.org/10.1038/s41573-024-00920-9.
Meier P, Legrand AJ, Adam D, Silke J. Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity. Nat Rev Cancer. 2024;24:299–315. https://doi.org/10.1038/s41568-024-00674-x.
Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. 2020;21:120–34. https://doi.org/10.1038/s41590-019-0561-4.
Alzeibak, R, Mishchenko, TA, Shilyagina, NY, Balalaeva, IV, Vedunova, MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J Immunother Cancer 2021;9. https://doi.org/10.1136/jitc-2020-001926.
Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. 2022;23:487–500. https://doi.org/10.1038/s41590-022-01132-2.
Petroni G, Galluzzi L. Impact of treatment schedule on the efficacy of cytostatic and immunostimulatory agents. Oncoimmunology. 2021;10:1889101. https://doi.org/10.1080/2162402x.2021.1889101.
Bravetti G, Falvo P, Talarico G, Orecchioni S, Bertolini F. Metronomic chemotherapy, dampening of immunosuppressive cells, antigen presenting cell activation, and T cells. A quartet against refractoriness and resistance to checkpoint inhibitors. Cancer Lett. 2023;577:216441. https://doi.org/10.1016/j.canlet.2023.216441.
Andre N, Tsai K, Carre M, Pasquier E. Metronomic Chemotherapy: Direct Targeting of Cancer Cells after all? Trends Cancer. 2017;3:319–25. https://doi.org/10.1016/j.trecan.2017.03.011.
Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016;6:446–59. https://doi.org/10.1158/2159-8290.Cd-15-0944.
Rios-Doria J, Durham N, Wetzel L, Rothstein R, Chesebrough J, Holoweckyj N, et al. Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia. 2015;17:661–70. https://doi.org/10.1016/j.neo.2015.08.004.
Chao Y, Liang C, Tao H, Du Y, Wu D, Dong Z, et al. Localized cocktail chemoimmunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. Sci Adv. 2020;6:eaaz4204. https://doi.org/10.1126/sciadv.aaz4204.
Fu D, Wu J, Lai J, Liu Y, Zhou L, Chen L, et al. T cell recruitment triggered by optimal dose platinum compounds contributes to the therapeutic efficacy of sequential PD-1 blockade in a mouse model of colon cancer. Am J Cancer Res. 2020;10:473–90.
Golchin S, Alimohammadi R, Rostami Nejad M, Jalali SA. Synergistic antitumor effect of anti-PD-L1 combined with oxaliplatin on a mouse tumor model. J Cell Physiol. 2019;234:19866–74. https://doi.org/10.1002/jcp.28585.
Zhou J, Yang T, Liu L, Lu B. Chemotherapy oxaliplatin sensitizes prostate cancer to immune checkpoint blockade therapies via stimulating tumor immunogenicity. Mol Med Rep. 2017;16:2868–74. https://doi.org/10.3892/mmr.2017.6908.
Park SJ, Ye W, Xiao R, Silvin C, Padget M, Hodge JW, et al. Cisplatin and oxaliplatin induce similar immunogenic changes in preclinical models of head and neck cancer. Oral Oncol. 2019;95:127–35. https://doi.org/10.1016/j.oraloncology.2019.06.016.
Zhao Z, Liu S, Sun R, Zhu W, Zhang Y, Liu T, et al. The combination of oxaliplatin and anti-PD-1 inhibitor promotes immune cells infiltration and enhances anti-tumor effect of PD-1 blockade in bladder cancer. Front Immunol. 2023;14:1085476. https://doi.org/10.3389/fimmu.2023.1085476.
Zhu H, Shan Y, Ge K, Lu J, Kong W, Jia C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell Oncol (Dordr). 2020;43:1203–14. https://doi.org/10.1007/s13402-020-00552-2.
Lévesque S, Le Naour J, Pietrocola F, Paillet J, Kremer M, Castoldi F, et al. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. Oncoimmunology. 2019;8:e1657375. https://doi.org/10.1080/2162402x.2019.1657375.
Iribarren K, Bloy N, Buqué A, Cremer I, Eggermont A, Fridman WH, et al. Trial Watch: Immunostimulation with Toll-like receptor agonists in cancer therapy. Oncoimmunology. 2016;5:e1088631. https://doi.org/10.1080/2162402x.2015.1088631.
Galluzzi L, Morselli E, Vicencio JM, Kepp O, Joza N, Tajeddine N, et al. Life, death and burial: multifaceted impact of autophagy. Biochem Soc Trans. 2008;36:786–90. https://doi.org/10.1042/bst0360786.
Vienot A, Pallandre JR, Renaude E, Viot J, Bouard A, Spehner L, et al. Chemokine switch regulated by TGF-β1 in cancer-associated fibroblast subsets determines the efficacy of chemo-immunotherapy. Oncoimmunology. 2022;11:2144669. https://doi.org/10.1080/2162402x.2022.2144669.
Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–58. https://doi.org/10.1038/onc.2010.500.
Luo R, Onyshchenko K, Wang L, Gaedicke S, Grosu AL, Firat E, et al. Necroptosis-dependent Immunogenicity of Cisplatin: Implications for Enhancing the Radiation-induced Abscopal Effect. Clin Cancer Res. 2023;29:667–83. https://doi.org/10.1158/1078-0432.Ccr-22-1591.
Liu P, Chen J, Zhao L, Hollebecque A, Kepp O, Zitvogel L, et al. PD-1 blockade synergizes with oxaliplatin-based, but not cisplatin-based, chemotherapy of gastric cancer. Oncoimmunology. 2022;11:2093518. https://doi.org/10.1080/2162402x.2022.2093518.
Principe N, Aston WJ, Hope DE, Tilsed CM, Fisher SA, Boon L, et al. Comprehensive Testing of Chemotherapy and Immune Checkpoint Blockade in Preclinical Cancer Models Identifies Additive Combinations. Front Immunol. 2022;13:872295. https://doi.org/10.3389/fimmu.2022.872295.
Nishimura J, Deguchi S, Tanaka H, Yamakoshi Y, Yoshii M, Tamura T, et al. Induction of Immunogenic Cell Death of Esophageal Squamous Cell Carcinoma by 5-Fluorouracil and Cisplatin. In Vivo. 2021;35:743–52. https://doi.org/10.21873/invivo.12315.
Yamazaki T, Buqué A, Ames TD, Galluzzi L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology. 2020;9:1721810. https://doi.org/10.1080/2162402x.2020.1721810.
Ding ZC, Lu X, Yu M, Lemos H, Huang L, Chandler P, et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014;74:3441–53. https://doi.org/10.1158/0008-5472.Can-13-3596.
Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, et al. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol. 2011;41:2977–86. https://doi.org/10.1002/eji.201141639.
Iida Y, Harashima N, Motoshima T, Komohara Y, Eto M, Harada M. Contrasting effects of cyclophosphamide on anti-CTL-associated protein 4 blockade therapy in two mouse tumor models. Cancer Sci. 2017;108:1974–84. https://doi.org/10.1111/cas.13337.
Choi Y, Kim SA, Jung H, Kim E, Kim YK, Kim S et al. Novel insights into paclitaxel's role on tumor-associated macrophages in enhancing PD-1 blockade in breast cancer treatment. J Immunother Cancer 2024;12. https://doi.org/10.1136/jitc-2024-008864.
Adkins I, Fucikova J, Garg AD, Agostinis P, Špíšek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology. 2014;3:e968434. https://doi.org/10.4161/21624011.2014.968434.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88. https://doi.org/10.1158/1078-0432.Ccr-09-0265.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618. https://doi.org/10.1038/ncomms15618.
Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020;21:1160–71. https://doi.org/10.1038/s41590-020-0751-0.
Barsoumian HB, Ramapriyan R, Younes AI, Caetano MS, Menon H, Comeaux NI et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J Immunother Cancer 2020;8. https://doi.org/10.1136/jitc-2020-000537.
Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11:728–34.
Pilones KA, Hensler M, Daviaud C, Kraynak J, Fucikova J, Galluzzi L, et al. Converging focal radiation and immunotherapy in a preclinical model of triple negative breast cancer: contribution of VISTA blockade. Oncoimmunology. 2020;9:1830524. https://doi.org/10.1080/2162402x.2020.1830524.
Newton JM, Hanoteau A, Liu HC, Gaspero A, Parikh F, Gartrell-Corrado RD, et al. Immune microenvironment modulation unmasks therapeutic benefit of radiotherapy and checkpoint inhibition. J Immunother Cancer. 2019;7:216. https://doi.org/10.1186/s40425-019-0698-6.
Wang L, Luo R, Onyshchenko K, Rao X, Wang M, Menz B et al. Adding liposomal doxorubicin enhances the abscopal effect induced by radiation/αPD1 therapy depending on tumor cell mitochondrial DNA and cGAS/STING. J Immunother Cancer 2023;11. https://doi.org/10.1136/jitc-2022-006235.
Vitale I, Yamazaki T, Wennerberg E, Sveinbjørnsson B, Rekdal Ø, Demaria S, et al. Targeting Cancer Heterogeneity with Immune Responses Driven by Oncolytic Peptides. Trends Cancer. 2021;7:557–72. https://doi.org/10.1016/j.trecan.2020.12.012.
Yamazaki T, Wennerberg E, Hensler M, Buqué A, Kraynak J, Fucikova J, et al. LTX-315-enabled, radiotherapy-boosted immunotherapeutic control of breast cancer by NK cells. Oncoimmunology. 2021;10:1962592. https://doi.org/10.1080/2162402x.2021.1962592.
Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 2012;72:3163–74. https://doi.org/10.1158/0008-5472.Can-12-0210.
Herrera FG, Ronet C, Ochoa de Olza M, Barras D, Crespo I, Andreatta M, et al. Low-Dose Radiotherapy Reverses Tumor Immune Desertification and Resistance to Immunotherapy. Cancer Discov. 2022;12:108–33. https://doi.org/10.1158/2159-8290.Cd-21-0003.
Zhou Y, Zhang W, Wang B, Wang P, Li D, Cao T et al. Mitochondria-targeted photodynamic therapy triggers GSDME-mediated pyroptosis and sensitizes anti-PD-1 therapy in colorectal cancer. J Immunother Cancer 2024;12. https://doi.org/10.1136/jitc-2023-008054.
Kaneko K, Acharya CR, Nagata H, Yang X, Hartman ZC, Hobeika A et al. Combination of a novel heat shock protein 90-targeted photodynamic therapy with PD-1/PD-L1 blockade induces potent systemic antitumor efficacy and abscopal effect against breast cancers. J Immunother Cancer 2022;10. https://doi.org/10.1136/jitc-2022-004793.
Arrieta VA, Gould A, Kim KS, Habashy KJ, Dmello C, Vázquez-Cervantes GI, et al. Ultrasound-mediated delivery of doxorubicin to the brain results in immune modulation and improved responses to PD-1 blockade in gliomas. Nat Commun. 2024;15:4698. https://doi.org/10.1038/s41467-024-48326-w.
Petroni G, Buqué A, Yamazaki T, Bloy N, Liberto MD, Chen-Kiang S, et al. Radiotherapy Delivered before CDK4/6 Inhibitors Mediates Superior Therapeutic Effects in ER(+) Breast Cancer. Clin Cancer Res. 2021;27:1855–63. https://doi.org/10.1158/1078-0432.Ccr-20-3871.
Wang W, Wu L, Zhang J, Wu H, Han E, Guo Q. Chemoimmunotherapy by combining oxaliplatin with immune checkpoint blockades reduced tumor burden in colorectal cancer animal model. Biochem Biophys Res Commun. 2017;487:1–7. https://doi.org/10.1016/j.bbrc.2016.12.180.
Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74:5458–68. https://doi.org/10.1158/0008-5472.Can-14-1258.
Paul S, Konig MF, Pardoll DM, Bettegowda C, Papadopoulos N, Wright KM, et al. Cancer therapy with antibodies. Nat Rev Cancer. 2024;24:399–426. https://doi.org/10.1038/s41568-024-00690-x.
Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A, et al. Immune checkpoint therapy-current perspectives and future directions. Cell. 2023;186:1652–69. https://doi.org/10.1016/j.cell.2023.03.006.
Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59. https://doi.org/10.1016/s1470-2045(19)30689-8.
Fabi A, Carbognin L, Botticelli A, Paris I, Fuso P, Savastano MC, et al. Real-world ANASTASE study of atezolizumab+nab-paclitaxel as first-line treatment of PD-L1-positive metastatic triple-negative breast cancer. NPJ Breast Cancer. 2023;9:73. https://doi.org/10.1038/s41523-023-00579-2.
Miles D, Gligorov J, André F, Cameron D, Schneeweiss A, Barrios C, et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann Oncol. 2021;32:994–1004. https://doi.org/10.1016/j.annonc.2021.05.801.
Cortes J, Rugo HS, Cescon DW, Im SA, Yusof MM, Gallardo C, et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2022;387:217–26. https://doi.org/10.1056/NEJMoa2202809.
Doki Y, Ajani JA, Kato K, Xu J, Wyrwicz L, Motoyama S, et al. Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. N Engl J Med. 2022;386:449–62. https://doi.org/10.1056/NEJMoa2111380.
Rha SY, Oh DY, Yañez P, Bai Y, Ryu MH, Lee J, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for HER2-negative advanced gastric cancer (KEYNOTE-859): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24:1181–95. https://doi.org/10.1016/s1470-2045(23)00515-6.
Janjigian YY, Ajani JA, Moehler M, Shen L, Garrido M, Gallardo C, et al. First-Line Nivolumab Plus Chemotherapy for Advanced Gastric, Gastroesophageal Junction, and Esophageal Adenocarcinoma: 3-Year Follow-Up of the Phase III CheckMate 649 Trial. J Clin Oncol. 2024;42:2012–20. https://doi.org/10.1200/jco.23.01601.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25:920–8. https://doi.org/10.1038/s41591-019-0432-4.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med. 2017;377:1919–29. https://doi.org/10.1056/NEJMoa1709937.
Spigel DR, Faivre-Finn C, Gray JE, Vicente D, Planchard D, Paz-Ares L, et al. Five-Year Survival Outcomes From the PACIFIC Trial: Durvalumab After Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. J Clin Oncol. 2022;40:1301–11. https://doi.org/10.1200/jco.21.01308.
Loibl S, Schneeweiss A, Huober J, Braun M, Rey J, Blohmer JU, et al. Neoadjuvant durvalumab improves survival in early triple-negative breast cancer independent of pathological complete response. Ann Oncol. 2022;33:1149–58. https://doi.org/10.1016/j.annonc.2022.07.1940.
Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med. 2020;382:810–21. https://doi.org/10.1056/NEJMoa1910549.
Loibl S, Untch M, Burchardi N, Huober J, Sinn BV, Blohmer JU, et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann Oncol. 2019;30:1279–88. https://doi.org/10.1093/annonc/mdz158.
Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med. 2022;386:1973–85. https://doi.org/10.1056/NEJMoa2202170.
Wakelee H, Liberman M, Kato T, Tsuboi M, Lee SH, Gao S, et al. Perioperative Pembrolizumab for Early-Stage Non-Small-Cell Lung Cancer. N Engl J Med. 2023;389:491–503. https://doi.org/10.1056/NEJMoa2302983.
Provencio M, Nadal E, González-Larriba JL, Martínez-Martí A, Bernabé R, Bosch-Barrera J, et al. Perioperative Nivolumab and Chemotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med. 2023;389:504–13. https://doi.org/10.1056/NEJMoa2215530.
Rozeman EA, Menzies AM, van Akkooi ACJ, Adhikari C, Bierman C, van de Wiel BA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 2019;20:948–60. https://doi.org/10.1016/s1470-2045(19)30151-2.
Rozeman EA, Hoefsmit EP, Reijers ILM, Saw RPM, Versluis JM, Krijgsman O, et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat Med. 2021;27:256–63. https://doi.org/10.1038/s41591-020-01211-7.
Reijers ILM, Menzies AM, van Akkooi ACJ, Versluis JM, van den Heuvel NMJ, Saw RPM, et al. Personalized response-directed surgery and adjuvant therapy after neoadjuvant ipilimumab and nivolumab in high-risk stage III melanoma: the PRADO trial. Nat Med. 2022;28:1178–88. https://doi.org/10.1038/s41591-022-01851-x.
Gross ND, Miller DM, Khushalani NI, Divi V, Ruiz ES, Lipson EJ, et al. Neoadjuvant Cemiplimab for Stage II to IV Cutaneous Squamous-Cell Carcinoma. N Engl J Med. 2022;387:1557–68. https://doi.org/10.1056/NEJMoa2209813.
Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26:566–76. https://doi.org/10.1038/s41591-020-0805-8.
Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N Engl J Med. 2022;386:2363–76. https://doi.org/10.1056/NEJMoa2201445.
Cubas R, Moskalenko M, Cheung J, Yang M, McNamara E, Xiong H, et al. Chemotherapy Combines Effectively with Anti-PD-L1 Treatment and Can Augment Antitumor Responses. J Immunol. 2018;201:2273–86. https://doi.org/10.4049/jimmunol.1800275.
Tanchot C, Terme M, Pere H, Tran T, Benhamouda N, Strioga M, et al. Tumor-infiltrating regulatory T cells: phenotype, role, mechanism of expansion in situ and clinical significance. Cancer Microenviron. 2013;6:147–57. https://doi.org/10.1007/s12307-012-0122-y.
Berckmans Y, Vankerckhoven A, Caro AA, Kempeneers J, Ceusters J, Thirion G et al. TIM3 Checkpoint Inhibition Fails to Prolong Survival in Ovarian Cancer-Bearing Mice. Cancers (Basel) 2024;16. https://doi.org/10.3390/cancers16061147.
Vitale I, Pietrocola F, Guilbaud E, Aaronson SA, Abrams JM, Adam D, et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023;30:1097–154. https://doi.org/10.1038/s41418-023-01153-w.
Jiménez-Cortegana C, Galassi C, Klapp V, Gabrilovich DI, Galluzzi L. Myeloid-Derived Suppressor Cells and Radiotherapy. Cancer Immunol Res. 2022;10:545–57. https://doi.org/10.1158/2326-6066.Cir-21-1105.
Reguera-Nuñez E, Xu P, Chow A, Man S, Hilberg F, Kerbel RS. Therapeutic impact of Nintedanib with paclitaxel and/or a PD-L1 antibody in preclinical models of orthotopic primary or metastatic triple negative breast cancer. J Exp Clin Cancer Res. 2019;38:16. https://doi.org/10.1186/s13046-018-0999-5.
Jotte R, Cappuzzo F, Vynnychenko I, Stroyakovskiy D, Rodríguez-Abreu D, Hussein M, et al. Atezolizumab in Combination With Carboplatin and Nab-Paclitaxel in Advanced Squamous NSCLC (IMpower131): Results From a Randomized Phase III Trial. J Thorac Oncol. 2020;15:1351–60. https://doi.org/10.1016/j.jtho.2020.03.028.
Nishio M, Barlesi F, West H, Ball S, Bordoni R, Cobo M, et al. Atezolizumab Plus Chemotherapy for First-Line Treatment of Nonsquamous NSCLC: Results From the Randomized Phase 3 IMpower132 Trial. J Thorac Oncol. 2021;16:653–64. https://doi.org/10.1016/j.jtho.2020.11.025.
Emens LA, Adams S, Barrios CH, Diéras V, Iwata H, Loi S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol. 2021;32:983–93. https://doi.org/10.1016/j.annonc.2021.05.355.
Govindan R, Szczesna A, Ahn MJ, Schneider CP, Gonzalez Mella PF, Barlesi F, et al. Phase III Trial of Ipilimumab Combined With Paclitaxel and Carboplatin in Advanced Squamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2017;35:3449–57. https://doi.org/10.1200/JCO.2016.71.7629.
Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W, et al. Phase III Randomized Trial of Ipilimumab Plus Etoposide and Platinum Versus Placebo Plus Etoposide and Platinum in Extensive-Stage Small-Cell Lung Cancer. J Clin Oncol. 2016;34:3740–8. https://doi.org/10.1200/jco.2016.67.6601.
Owonikoko TK, Park K, Govindan R, Ready N, Reck M, Peters S, et al. Nivolumab and Ipilimumab as Maintenance Therapy in Extensive-Disease Small-Cell Lung Cancer: CheckMate 451. J Clin Oncol. 2021;39:1349–59. https://doi.org/10.1200/jco.20.02212.
Spigel DR, Vicente D, Ciuleanu TE, Gettinger S, Peters S, Horn L, et al. Second-line nivolumab in relapsed small-cell lung cancer: CheckMate 331(✩). Ann Oncol. 2021;32:631–41. https://doi.org/10.1016/j.annonc.2021.01.071.
Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW, et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6:1571–80. https://doi.org/10.1001/jamaoncol.2020.3370.
Chao J, Fuchs CS, Shitara K, Tabernero J, Muro K, Van Cutsem E, et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol. 2021;7:895–902. https://doi.org/10.1001/jamaoncol.2021.0275.
Lenz HJ, Parikh A, Spigel DR, Cohn AL, Yoshino T, Kochenderfer M et al. (2024) Modified FOLFOX6 plus bevacizumab with and without nivolumab for first-line treatment of metastatic colorectal cancer: phase 2 results from the CheckMate 9X8 randomized clinical trial. J Immunother Cancer 2024;12. https://doi.org/10.1136/jitc-2023-008409.
Lenz H-J, Parikh AR, Spigel DR, Cohn AL, Yoshino T, Kochenderfer MD, et al. Nivolumab (NIVO) + 5-fluorouracil/leucovorin/oxaliplatin (mFOLFOX6)/bevacizumab (BEV) versus mFOLFOX6/BEV for first-line (1L) treatment of metastatic colorectal cancer (mCRC): Phase 2 results from CheckMate 9X8. Journal of Clinical Oncology. 2022;40:8–8. https://doi.org/10.1200/JCO.2022.40.4_suppl.008.
Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24:1935–49. https://doi.org/10.1093/neuonc/noac116.
Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023;25:123–34. https://doi.org/10.1093/neuonc/noac099.
Petrylak DP, Ratta R, Matsubara N, Korbenfeld EP, Gafanov R, Mourey L, et al. Pembrolizumab plus docetaxel for patients with metastatic castration-resistant prostate cancer (mCRPC): Randomized, double-blind, phase 3 KEYNOTE-921 study. Journal of Clinical Oncology. 2023;41:19–19. https://doi.org/10.1200/JCO.2023.41.6_suppl.19.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12. https://doi.org/10.1016/s1470-2045(14)70189-5.
Powles T, Csőszi T, Özgüroğlu M, Matsubara N, Géczi L, Cheng SY, et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:931–45. https://doi.org/10.1016/s1470-2045(21)00152-2.
Grande E, Arranz J, De Santis M, Bamias A, Kikuchi E, Del Muro XG, et al. Atezolizumab plus chemotherapy versus placebo plus chemotherapy in untreated locally advanced or metastatic urothelial carcinoma (IMvigor130): final overall survival analysis results from a randomised, controlled, phase 3 study. Lancet Oncol. 2024;25:29–45. https://doi.org/10.1016/s1470-2045(23)00540-5.
Lee NY, Ferris RL, Psyrri A, Haddad RI, Tahara M, Bourhis J, et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2021;22:450–62. https://doi.org/10.1016/s1470-2045(20)30737-3.
Bourhis J, Tao Y, Sun X, Sire C, Martin L, Liem X, et al. Avelumab-cetuximab-radiotherapy versus standards of care in patients with locally advanced squamous cell carcinoma of head and neck (LA-SCCHN): Randomized phase III GORTEC-REACH trial. Annals of Oncology. 2021;32:S1310–S1310. https://doi.org/10.1016/j.annonc.2021.08.2112.
Goodman RS, Johnson DB, Balko JM. Corticosteroids and Cancer Immunotherapy. Clin Cancer Res. 2023;29:2580–7. https://doi.org/10.1158/1078-0432.Ccr-22-3181.
Rached L, Laparra A, Sakkal M, Danlos FX, Barlesi F, Carbonnel F, et al. Toxicity of immunotherapy combinations with chemotherapy across tumor indications: Current knowledge and practical recommendations. Cancer Treat Rev. 2024;127:102751. https://doi.org/10.1016/j.ctrv.2024.102751.
Gianni L, Huang CS, Egle D, Bermejo B, Zamagni C, Thill M, et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple-negative, early high-risk and locally advanced breast cancer: NeoTRIP Michelangelo randomized study. Ann Oncol. 2022;33:534–43. https://doi.org/10.1016/j.annonc.2022.02.004.
Huober J, Barrios CH, Niikura N, Jarząb M, Chang YC, Huggins-Puhalla SL, et al. Atezolizumab With Neoadjuvant Anti-Human Epidermal Growth Factor Receptor 2 Therapy and Chemotherapy in Human Epidermal Growth Factor Receptor 2-Positive Early Breast Cancer: Primary Results of the Randomized Phase III IMpassion050 Trial. J Clin Oncol. 2022;40:2946–56. https://doi.org/10.1200/jco.21.02772.
Pignata S, Bookman M, Sehouli J, Miller A, Penson RT, Taskiran C, et al. Overall survival and patient-reported outcome results from the placebo-controlled randomized phase III IMagyn050/GOG 3015/ENGOT-OV39 trial of atezolizumab for newly diagnosed stage III/IV ovarian cancer. Gynecol Oncol. 2023;177:20–31. https://doi.org/10.1016/j.ygyno.2023.06.018.
Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer 2020;8. https://doi.org/10.1136/jitc-2019-000337.
Doloff JC, Chen CS, Waxman DJ. Anti-tumor innate immunity activated by intermittent metronomic cyclophosphamide treatment of 9L brain tumor xenografts is preserved by anti-angiogenic drugs that spare VEGF receptor 2. Mol Cancer. 2014;13:158. https://doi.org/10.1186/1476-4598-13-158.
Maharjan R, Choi JU, Kweon S, Pangeni R, Lee NK, Park SJ, et al. A novel oral metronomic chemotherapy provokes tumor specific immunity resulting in colon cancer eradication in combination with anti-PD-1 therapy. Biomaterials. 2022;281:121334. https://doi.org/10.1016/j.biomaterials.2021.121334.
Park SJ, Kweon S, Moyo MK, Kim HR, Choi JU, Lee NK, et al. Immune modulation of the liver metastatic colorectal cancer microenvironment via the oral CAPOX-mediated cGAS-STING pathway. Biomaterials. 2024;310:122625. https://doi.org/10.1016/j.biomaterials.2024.122625.
Wu J, Waxman DJ. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018;419:210–21. https://doi.org/10.1016/j.canlet.2018.01.050.
Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394:1929–39. https://doi.org/10.1016/s0140-6736(19)32222-6.
Ni W, Xiao Z, Zhou Z, Chen D, Feng Q, Liang J, et al. Severe radiation-induced lymphopenia during postoperative radiotherapy or chemoradiotherapy has poor prognosis in patients with stage IIB-III after radical esophagectomy: A post hoc analysis of a randomized controlled trial. Front Oncol. 2022;12:936684. https://doi.org/10.3389/fonc.2022.936684.
Smith RE. Trends in recommendations for myelosuppressive chemotherapy for the treatment of solid tumors. J Natl Compr Canc Netw. 2006;4:649–58. https://doi.org/10.6004/jnccn.2006.0056.
Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia: risks, consequences, and new directions for its management. Cancer. 2004;100:228–37. https://doi.org/10.1002/cncr.11882.
Carey PJ. Drug-induced myelosuppression : diagnosis and management. Drug Saf. 2003;26:691–706. https://doi.org/10.2165/00002018-200326100-00003.
Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021;27:212–24. https://doi.org/10.1038/s41591-021-01233-9.
Sanmamed MF, Chester C, Melero I, Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann Oncol. 2016;27:1190–8. https://doi.org/10.1093/annonc/mdw041.
Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018;8:1358–65. https://doi.org/10.1158/2159-8290.Cd-18-0044.
Buque A, Bloy N, Perez-Lanzon M, Iribarren K, Humeau J, Pol JG, et al. Immunoprophylactic and immunotherapeutic control of hormone receptor-positive breast cancer. Nat Commun. 2020;11:3819. https://doi.org/10.1038/s41467-020-17644-0.
Buque A, Galluzzi L. Modeling Tumor Immunology and Immunotherapy in Mice. Trends Cancer. 2018;4:599–601. https://doi.org/10.1016/j.trecan.2018.07.003.
Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med. 2017;9:137–53. https://doi.org/10.15252/emmm.201606857.
Kroemer G, Chan TA, Eggermont AMM, Galluzzi L. Immunosurveillance in clinical cancer management. CA Cancer J Clin. 2024;74:187–202. https://doi.org/10.3322/caac.21818.
Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013. https://doi.org/10.1038/s41419-020-03221-2.
Fonseca C, Dranoff G. Capitalizing on the immunogenicity of dying tumor cells. Clin Cancer Res. 2008;14:1603–8. https://doi.org/10.1158/1078-0432.Ccr-07-2245.
Workenhe ST, Pol J, Kroemer G. Tumor-intrinsic determinants of immunogenic cell death modalities. Oncoimmunology. 2021;10:1893466. https://doi.org/10.1080/2162402x.2021.1893466.
Galassi C, Chan TA, Vitale I, Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. 2024. https://doi.org/10.1016/j.ccell.2024.09.010.
Fucikova J, Moserova I, Urbanova L, Bezu L, Kepp O, Cremer I, et al. Prognostic and Predictive Value of DAMPs and DAMP-Associated Processes in Cancer. Front Immunol. 2015;6:402. https://doi.org/10.3389/fimmu.2015.00402.
Zitvogel L, Fidelle M, Kroemer G. Long-distance microbial mechanisms impacting cancer immunosurveillance. Immunity. 2024;57:2013–29. https://doi.org/10.1016/j.immuni.2024.07.020.
Montégut L, de Cabo R, Zitvogel L, Kroemer G. Science-Driven Nutritional Interventions for the Prevention and Treatment of Cancer. Cancer Discov. 2022;12:2258–79. https://doi.org/10.1158/2159-8290.Cd-22-0504.
Shaikh SR, Beck MA, Alwarawrah Y, MacIver NJ. Emerging mechanisms of obesity-associated immune dysfunction. Nat Rev Endocrinol. 2024;20:136–48. https://doi.org/10.1038/s41574-023-00932-2.
Cortellini A, Tucci M, Adamo V, Stucci, LS, Russo A, Tanda ET et al. (2020) Integrated analysis of concomitant medications and oncological outcomes from PD-1/PD-L1 checkpoint inhibitors in clinical practice. J Immunother Cancer 2020;8. https://doi.org/10.1136/jitc-2020-001361.
Thompson ME, Highley MS. Interaction between paclitaxel and warfarin. Ann Oncol. 2003;14:500. https://doi.org/10.1093/annonc/mdg096.
Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. https://doi.org/10.1038/cdd.2014.137.
Efimova I, Catanzaro E, Van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer 2020;8. https://doi.org/10.1136/jitc-2020-001369.
Catanzaro E, Demuynck R, Naessens F, Galluzzi L, Krysko DV. Immunogenicity of ferroptosis in cancer: a matter of context? Trends Cancer. 2024;10:407–16. https://doi.org/10.1016/j.trecan.2024.01.013.
Chen J, Larsson L, Swarbrick A, Lundeberg J. Spatial landscapes of cancers: insights and opportunities. Nat Rev Clin Oncol. 2024;21:660–74. https://doi.org/10.1038/s41571-024-00926-7.
Tivey A, Church M, Rothwell D, Dive C, Cook N. Circulating tumour DNA - looking beyond the blood. Nat Rev Clin Oncol. 2022;19:600–12. https://doi.org/10.1038/s41571-022-00660-y.
Clasen K, Welz S, Faltin H, Zips D, Eckert F. Dynamics of HMBG1 (High Mobility Group Box 1) during radiochemotherapy correlate with outcome of HNSCC patients. Strahlenther Onkol. 2022;198:194–200. https://doi.org/10.1007/s00066-021-01860-8.
Exner R, Sachet M, Arnold T, Zinn-Zinnenburg M, Michlmayr A, Dubsky P, et al. Prognostic value of HMGB1 in early breast cancer patients under neoadjuvant chemotherapy. Cancer Med. 2016;5:2350–8. https://doi.org/10.1002/cam4.827.
Loi S, Curigliano G, Salgado RF, Romero Diaz RI, Delaloge S, Rojas C, et al. LBA20 A randomized, double-blind trial of nivolumab (NIVO) vs placebo (PBO) with neoadjuvant chemotherapy (NACT) followed by adjuvant endocrine therapy (ET) ± NIVO in patients (pts) with high-risk, ER+ HER2− primary breast cancer (BC). Annals of Oncology. 2023;34:S1259–S1260. https://doi.org/10.1016/j.annonc.2023.10.010.
Monk BJ, Colombo N, Tewari KS, Dubot C, Caceres MV, Hasegawa K, et al. First-Line Pembrolizumab + Chemotherapy Versus Placebo + Chemotherapy for Persistent, Recurrent, or Metastatic Cervical Cancer: Final Overall Survival Results of KEYNOTE-826. J Clin Oncol. 2023;41:5505–11. https://doi.org/10.1200/jco.23.00914.
Makker V, Colombo N, Casado Herráez A, Santin AD, Colomba E, Miller DS, et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N Engl J Med. 2022;386:437–48. https://doi.org/10.1056/NEJMoa2108330.
Sun JM, Shen L, Shah MA, Enzinger P, Adenis A, Doi T, et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): a randomised, placebo-controlled, phase 3 study. Lancet. 2021;398:759–71. https://doi.org/10.1016/s0140-6736(21)01234-4.
Shah MA, Sun J-M, Shen L, Kato K, Enzinger PC, Adenis A, et al. First-line pembrolizumab (pembro) plus chemotherapy (chemo) for advanced esophageal cancer: 5-year outcomes from the phase 3 KEYNOTE-590 study. Journal of Clinical Oncology. 2024;42:250–250. https://doi.org/10.1200/JCO.2024.42.3_suppl.250.
Xu J, Jiang H, Pan Y, Gu K, Cang S, Han L, et al. Sintilimab Plus Chemotherapy for Unresectable Gastric or Gastroesophageal Junction Cancer: The ORIENT-16 Randomized Clinical Trial. Jama. 2023;330:2064–74. https://doi.org/10.1001/jama.2023.19918.
Qiu MZ, Oh DY, Kato K, Arkenau T, Tabernero J, Correa MC, et al. Tislelizumab plus chemotherapy versus placebo plus chemotherapy as first line treatment for advanced gastric or gastro-oesophageal junction adenocarcinoma: RATIONALE-305 randomised, double blind, phase 3 trial. Bmj. 2024;385:e078876. https://doi.org/10.1136/bmj-2023-078876.
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27–40. https://doi.org/10.1016/s0140-6736(21)00797-2.
Janjigian YY, Kawazoe A, Yañez P, Li N, Lonardi S, Kolesnik O, et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature. 2021;600:727–30. https://doi.org/10.1038/s41586-021-04161-3.
Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr., et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915–28. https://doi.org/10.1016/s0140-6736(19)32591-7.
Harrington KJ, Burtness B, Greil R, Soulières D, Tahara M, de Castro G Jr., et al. Pembrolizumab With or Without Chemotherapy in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: Updated Results of the Phase III KEYNOTE-048 Study. J Clin Oncol. 2023;41:790–802. https://doi.org/10.1200/jco.21.02508.
Dzienis M, Cundom J, Fuentes CS, Spreafico A, Nordlinger M, Pastor AV, et al. Pembrolizumab Plus Carboplatin and Paclitaxel as First-Line Therapy for Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (KEYNOTE-B10): A Single-Arm Phase IV Trial. J Clin Oncol. 2024;42:2989–99. https://doi.org/10.1200/jco.23.02625.
Sugawara S, Lee JS, Kang JH, Kim HR, Inui N, Hida T, et al. Nivolumab with carboplatin, paclitaxel, and bevacizumab for first-line treatment of advanced nonsquamous non-small-cell lung cancer. Ann Oncol. 2021;32:1137–47. https://doi.org/10.1016/j.annonc.2021.06.004.
Kim HR, Sugawara S, Lee JS, Kang JH, Inui N, Hida T, et al. First-line nivolumab, paclitaxel, carboplatin, and bevacizumab for advanced non-squamous non-small cell lung cancer: Updated survival analysis of the ONO-4538-52/TASUKI-52 randomized controlled trial. Cancer Med. 2023;12:17061–7. https://doi.org/10.1002/cam4.6348.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301. https://doi.org/10.1056/NEJMoa1716948.
Socinski MA, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, et al. IMpower150 Final Overall Survival Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in First-Line Metastatic Nonsquamous NSCLC. J Thorac Oncol. 2021;16:1909–24. https://doi.org/10.1016/j.jtho.2021.07.009.
Cheng Y, Yang JC, Okamoto I, Zhang L, Hu J, Wang D, et al. Pembrolizumab plus chemotherapy for advanced non-small-cell lung cancer without tumor PD-L1 expression in Asia. Immunotherapy. 2023;15:1029–44. https://doi.org/10.2217/imt-2023-0043.
Gadgeel SM, Rodríguez-Abreu D, Halmos B, Garassino MC, Kurata T, Cheng Y, et al. Pembrolizumab Plus Chemotherapy for Metastatic NSCLC With Programmed Cell Death Ligand 1 Tumor Proportion Score Less Than 1%: Pooled Analysis of Outcomes After Five Years of Follow-Up. J Thorac Oncol. 2024;19:1228–41. https://doi.org/10.1016/j.jtho.2024.04.011.
West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:924–37. https://doi.org/10.1016/s1470-2045(19)30167-6.
Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:198–211. https://doi.org/10.1016/s1470-2045(20)30641-0.
Reck M, Ciuleanu TE, Schenker M, Bordenave S, Cobo M, Juan-Vidal O, et al. Five-year outcomes with first-line nivolumab plus ipilimumab with 2 cycles of chemotherapy versus 4 cycles of chemotherapy alone in patients with metastatic non-small cell lung cancer in the randomized CheckMate 9LA trial. Eur J Cancer. 2024;211:114296. https://doi.org/10.1016/j.ejca.2024.114296.
Cheng Y, Zhang L, Hu J, Wang D, Hu C, Zhou J, et al. Pembrolizumab Plus Chemotherapy for Chinese Patients With Metastatic Squamous NSCLC in KEYNOTE-407. JTO Clin Res Rep. 2021;2:100225. https://doi.org/10.1016/j.jtocrr.2021.100225.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379:2040–51. https://doi.org/10.1056/NEJMoa1810865.
Novello S, Kowalski DM, Luft A, Gümüş M, Vicente D, Mazières J, et al. Pembrolizumab Plus Chemotherapy in Squamous Non-Small-Cell Lung Cancer: 5-Year Update of the Phase III KEYNOTE-407 Study. J Clin Oncol. 2023;41:1999–2006. https://doi.org/10.1200/jco.22.01990.
Rudin CM, Awad MM, Navarro A, Gottfried M, Peters S, Csőszi T, et al. Pembrolizumab or Placebo Plus Etoposide and Platinum as First-Line Therapy for Extensive-Stage Small-Cell Lung Cancer: Randomized, Double-Blind, Phase III KEYNOTE-604 Study. J Clin Oncol. 2020;38:2369–79. https://doi.org/10.1200/jco.20.00793.
Kim HR, Awad MM, Navarro A, Gottfried M, Peters S, Csőszi T, et al. Patient-Reported Health-Related Quality of Life in KEYNOTE-604: Pembrolizumab or Placebo Added to Etoposide and Platinum as First-Line Therapy for Extensive-Stage SCLC. JTO Clin Res Rep. 2023;4:100572. https://doi.org/10.1016/j.jtocrr.2023.100572.
Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018;379:2220–9. https://doi.org/10.1056/NEJMoa1809064.
Nishio M, Sugawara S, Atagi S, Akamatsu H, Sakai H, Okamoto I, et al. Subgroup Analysis of Japanese Patients in a Phase III Study of Atezolizumab in Extensive-stage Small-cell Lung Cancer (IMpower133). Clin Lung Cancer. 2019;20:469–76.e461. https://doi.org/10.1016/j.cllc.2019.07.005.
Liu SV, Reck M, Mansfield AS, Mok T, Scherpereel A, Reinmuth N, et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients With Extensive-Stage Small-Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). J Clin Oncol. 2021;39:619–30. https://doi.org/10.1200/jco.20.01055.
Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396:1090–1100. https://doi.org/10.1016/s0140-6736(20)31953-x.
Schmid P, Cortes J, Dent R, Pusztai L, McArthur H, Kümmel S, et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N Engl J Med. 2022;386:556–67. https://doi.org/10.1056/NEJMoa2112651.
Lassman AB, Polley MC, Iwamoto FM, Sloan AE, Wang TJC, Aldape KD, et al. Pl02.3.a NRG oncology study bn007: randomized phase ii/iii trial of ipilimiumab (ipi) plus nivolumab (NIVO) vs. temozolomide (TMZ) in MGMT-unmethylated (UMGMT) newly diagnosed glioblastoma (NGBM). Neuro-Oncology. 2023;25:ii2–ii2. https://doi.org/10.1093/neuonc/noad137.005.
Mell LK, Torres-Saavedra P, Wong S, Chang S, Kish JA, Minn AJ, et al. Radiotherapy with Durvalumab vs. Cetuximab in Patients with Locoregionally Advanced Head and Neck Cancer and a Contraindication to Cisplatin: Phase II Results of NRG-HN004. International Journal of Radiation Oncology*Biology*Physics. 2022;114:1058. https://doi.org/10.1016/j.ijrobp.2022.09.003.
Machiels JP, Tao Y, Licitra L, Burtness B, Tahara M, Rischin D, et al. Pembrolizumab plus concurrent chemoradiotherapy versus placebo plus concurrent chemoradiotherapy in patients with locally advanced squamous cell carcinoma of the head and neck (KEYNOTE-412): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2024;25:572–87. https://doi.org/10.1016/s1470-2045(24)00100-1.
Moore KN, Bookman M, Sehouli J, Miller A, Anderson C, Scambia G, et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J Clin Oncol. 2021;39:1842–55. https://doi.org/10.1200/jco.21.00306.
Monk BJ, Colombo N, Oza AM, Fujiwara K, Birrer MJ, Randall L, et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22:1275–89. https://doi.org/10.1016/s1470-2045(21)00342-9.
Pujade-Lauraine E, Fujiwara K, Ledermann JA, Oza AM, Kristeleit R, Ray-Coquard IL, et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): an open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 2021;22:1034–46. https://doi.org/10.1016/s1470-2045(21)00216-3.
Bamias A, Davis ID, Galsky MD, Arranz J, Kikuchi E, Grande E, et al. Atezolizumab monotherapy versus chemotherapy in untreated locally advanced or metastatic urothelial carcinoma (IMvigor130): final overall survival analysis from a randomised, controlled, phase 3 study. Lancet Oncol. 2024;25:46–61. https://doi.org/10.1016/s1470-2045(23)00539-9.
Wolf NK, Kissiov DU, Raulet DH. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat Rev Immunol. 2023;23:90–105. https://doi.org/10.1038/s41577-022-00732-1.
Mensurado S, Blanco-Domínguez R, Silva-Santos B. The emerging roles of γδ T cells in cancer immunotherapy. Nat Rev Clin Oncol. 2023;20:178–91. https://doi.org/10.1038/s41571-022-00722-1.
Gebhardt T, Park SL, Parish IA. Stem-like exhausted and memory CD8(+) T cells in cancer. Nat Rev Cancer. 2023;23:780–98. https://doi.org/10.1038/s41568-023-00615-0.
Holder AM, Dedeilia A, Sierra-Davidson K, Cohen S, Liu D, Parikh A, et al. Defining clinically useful biomarkers of immune checkpoint inhibitors in solid tumours. Nat Rev Cancer. 2024;24:498–512. https://doi.org/10.1038/s41568-024-00705-7.
Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. 2022;19:254–67. https://doi.org/10.1038/s41571-022-00600-w.
Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020;6:38. https://doi.org/10.1038/s41572-020-0160-6.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75. https://doi.org/10.1038/nrc3380.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. https://doi.org/10.1038/nri.2016.107.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701. https://doi.org/10.1084/jem.20050915.
Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016;15:274–87. https://doi.org/10.1016/j.celrep.2016.03.037.
Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reise Sousa C, et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8⁺ T cells. Science. 2015;350:328–34. https://doi.org/10.1126/science.aad0395.
Li X, Li Y, Tuerxun H, Zhao Y, Liu X, Zhao Y. Firing up “cold” tumors: Ferroptosis causes immune activation by improving T cell infiltration. Biomed Pharmacother. 2024;179:117298. https://doi.org/10.1016/j.biopha.2024.117298.
Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676. https://doi.org/10.1038/s41467-022-31218-2.
Mishchenko T, Balalaeva I, Gorokhova A, Vedunova M, Krysko DV. Which cell death modality wins the contest for photodynamic therapy of cancer? Cell Death Dis. 2022;13:455. https://doi.org/10.1038/s41419-022-04851-4.
Acknowledgements
LG is supported (as a PI unless otherwise indicated) by one R01 grant from the NIH/NCI (#CA271915), by two Breakthrough Level 2 grants from the US DoD BCRP (#BC180476P1, #BC210945), by a grant from the STARR Cancer Consortium (#I16--0064), by a Transformative Breast Cancer Consortium Grant from the US DoD BCRP (#W81XWH2120034, PI: Formenti), by a U54 grant from NIH/NCI (#CA274291) from the Leukemia and Lymphoma Society (LLS), by the 2019 Laura Ziskin Prize in Translational Research (#ZP--6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by a pre-SPORE grant (PI: Demaria, Formenti) and a Clinical Trials Innovation Grant from the Sandra and Edward Meyer of Radiation Oncology at Weill Cornell Medicine (New York, US) and Fox Chase Cancer Center (Philadelphia, US), by industrial collaborations with Lytix Biopharma (Oslo, Norway), Promontory (New York, US) and Onxeo (Paris, France), as well as by donations from Promontory (New York, US), the Luke Heller TECPR2 Foundation (Boston, US), Sotio a.s. (Prague, Czech Republic), Lytix Biopharma (Oslo, Norway), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy), and Noxopharm (Chatswood, Australia). DVK is supported by the Flanders Research Foundation (FWO) and the Belgian National Fund for Scientific Research (F.R.S.-FNRS) under the Excellence of Science (EOS) program (#40007488), FWO grant (#G016221N), two Special Research Fund (BOF) grants from Ghent University (#BOF/IOP/2022/033, #BOF23/GOA/029), IOF “PULSE” grant (#F2023/IOF- ConcepTT/033) and IOF “IMMUNO-FER-GUARD” grant (#F2023/IOF-ConcepTT/106) from Ghent University. EC holds a Marie Sklodowska Curie (MSCA) postdoctoral fellowship (HORIZON-MSCA-2022 PF-01).
Author information
Authors and Affiliations
Contributions
LG and DVK conceived the article. EC wrote the first version of the manuscript with constructive input from LG and DVK. MBV and EC prepared the display items under supervision from LG and DVK. All the authors approved the submitted version of the article.
Corresponding authors
Ethics declarations
Competing interests
LG holds research contracts with Lytix Biopharma, Promontory and Onxeo; Has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, AbbVie, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Imvax, Sotio, Promontory, Noxopharm, EduCom, and the Luke Heller TECPR2 Foundation; and holds Promontory stock options. The other authors have no conflicts of interest to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Catanzaro, E., Beltrán-Visiedo, M., Galluzzi, L. et al. Immunogenicity of cell death and cancer immunotherapy with immune checkpoint inhibitors. Cell Mol Immunol 22, 24–39 (2025). https://doi.org/10.1038/s41423-024-01245-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-024-01245-8
Keywords
This article is cited by
-
Identification and validation of CDK1 as a promising therapeutic target for Eriocitrin in colorectal cancer: a combined bioinformatics and experimental approach
BMC Cancer (2025)
-
Translational selenium nanomedicine synergizes with nab-paclitaxel to enhance antitumor effects in esophageal squamous cell cancer via selenoprotein N-mediated ER stress
Journal of Nanobiotechnology (2025)
-
Bionic gene delivery system activates tumor autophagy and immunosuppressive niche to sensitize anti-PD-1 treatment against STK11-mutated lung adenocarcinoma
Journal of Nanobiotechnology (2025)
-
Aggregation induced emission luminogen bacteria hybrid bionic robot for multimodal phototheranostics and immunotherapy
Nature Communications (2025)
