
Abstract
Cancer cells secrete extracellular vesicles (EV) encapsulating bioactive cargoes to facilitate inter-organ communication in vivo and are emerging as critical mediators of tumor progression and metastasis, a condition which is often accompanied by a dysregulated cholesterol metabolism. Whether EVs are involved in the control of cholesterol homeostasis during tumor metastasis is still undefined and warrant further investigation. Here, we find that breast cancer-derived exosomal miR-9-5p induces the expression of HMGCR and CH25H, two enzymes involved in cholesterol synthesis and the conversion of 25-hydroxycholesterol from cholesterol by targeting INSIG1, INSIG2 and ATF3 genes in the liver. Notably, in vivo miR-9-5p antagomir treatment and genetic CH25H ablation prevents tumor metastasis in a mouse model of breast cancer. Thus, our findings reveal the regulatory mechanism of tumor-derived miR-9-5p in liver metastasis by linking oxysterol metabolism and Kupffer cell polarization, shedding light on future applications for cancer diagnosis and treatment.
Similar content being viewed by others


Introduction
Breast cancer (BC) is the most commonly diagnosed cancer type worldwide1,2. Most breast cancer-related deaths result from metastasis to distant organs, such as the brain, bone, lung and liver3,4. There are distinct alterations in gene expression, immune signatures or metabolic profiling underlie metastasis to the specific metastatic sites5. Before the formation of micro- and macro-metastasis, disseminated cancer cells need to overcome sequential inhibitory signals, survival in circulation, arrest at the distant site and extravasation6,7. Complicated factors residing in tumor cells or tumor microenvironment mediate tumor invasion and progressive outgrowth at a distant site8. For instance, immune cells and their mediators are known to facilitate BC metastasis and cancer cell dissemination is highly correlated with the capability to evade immunosurveillance8,9,10. Besides, liver-metastatic BC cells exhibit a unique metabolic program compared to the bone- or lung-metastatic cells, characterized by an increased glycolysis rate but a concomitant reduction in mitochondrial metabolism11. Therefore, to cope with complicated challenges during the metastatic process, tumor cells dynamically take multiple strategies to induce vascular leakage, recruitment of pro-angiogenic immune cells, or influence of organotropism to extend the effect of the intrinsic factors, such as the secretion of nanoparticles12,13.
Cancer exerts systemic effects partially through secreting extracellular vesicles (EV) that travel throughout the body via blood circulation and deliver bioactive cargo including miRNAs, lipids and proteins to various organs14,15,16. The biological function of those circulating EVs was involved in reprogramming cells in the tumor microenvironment or in a metastatic niche to promote tumor growth, immune evasion, and metastasis15,17,18,19. Indeed, cancer patients-secreted-circulating EVs (exosomes and micro-vesicles) have been associated with metastasis or relapse, serving as important diagnostic and prognostic markers as well as therapeutic targets20. Moreover, tumor-secreted EVs are major mediators for cancer cells to adapt and modify foreign microenvironments21. Accumulating evidence demonstrates that microRNA as well as proteins could be transferred to local or distant cells in these secretory forms to modulate cell function, emerging as a new group of messengers and effectors between intercellular communication22.
Dysregulated lipid metabolism has been reported to regulate cancer incidence and metastasis23,24. Among the environmental factors believed to be responsible for this phenomenon, cholesterol has recently received considerable attention. Cholesterol is a precursor of bile acids, steroid hormones and vitamin D, and a component of cell membranes, which plays a crucial role in cell growth and differentiation25,26. Upregulated cholesterol biosynthesis has been discovered in several types of cancers, where it supports the growth, metastasis, stemness, and therapeutic resistance of tumors27. In BC, cholesterol and its metabolites have been found to promote tumor progression both preclinically and clinically28. Compared to other BC subtypes, TNBC displays elevated cholesterol biosynthesis, which could have profound biological functions and indicate potential therapeutic strategies29. However, the regulatory mechanism and biological function of BC-derived EVs in cholesterol homeostasis during tumor metastasis remain poorly defined. Here, we show that the miRNA cargo loaded by cancer-secreted extracellular vesicles, functioning as “tri-brakes” (targeting three genes on the cholesterol homeostasis pathway), promotes cholesterol conversion into 25-hydroxycholesterol in liver. This onco-metabolite thereby induces breast cancer liver metastasis. This important result provides a new understanding of the dynamic interaction between cancer and host at the systemic level, providing valuable insights into how cancer alters specific metabolite level, which we consider to be highly innovative. The study also reveals the possibility of using circulating miR-9-5p and 25-hydroxycholesterol as novel blood-based biomarkers to identify breast cancer patients with high risk of developing metastasis to guide preventive screenings and early intervention.
Results
Breast cancer-derived EV enhanced cholesterol biosynthesis in liver
To explore the systemic effect of breast cancer-derived EVs in vivo, both human triple-negative breast cancer MDA-MB-231 cells and MDA-MB-231/Rab27a KD cells with stable expression of a membrane-targeted Lck-GFP were used to establish orthotopic xenograft tumors (231/Ctrl and 231/Rab27a KD) through injection into the mammary fat pads (Fig. 1a). Notably, a previous report showed Rab27a was involved in EV secretion and cells with Rab27a knockdown displayed impaired EV secretion30. Both MDA-MB-231 EV and 231/Rab27a KD EV displayed classic exosome size distribution (Supplementary Fig. 1a). In agreement with our previous finding18, decreased Rab27a expression in MDA-MB-231 cells leads to significantly reduced EV secretion (Supplementary Fig. 1b, c). We performed nanoflow cytometry analysis to further confirm the presence of CD63 and GFP proteins (~20% expression) on the EV membrane (Supplementary Fig. 1d). We detected GFP signals in the liver of 231/Ctrl mice rather than tumor free mice or 231/Rab27a KD mice, indicating uptake of GFP-labeled EVs by the liver from circulation in vivo (Fig. 1a). We thereby performed RNA-sequencing for the liver tissues, and the following GSEA enrichment analysis revealed the cholesterol biosynthesis pathway was the only significantly upregulated pathway in 231/Ctrl mice than either tumor free or 231/Rab27a KD mice (Fig. 1b). Furthermore, the total cholesterol level (TC) was increased in both serum and liver tissues from 231/Ctrl mice than the control groups (Fig. 1c). Consistently, there was more lipid accumulation in the liver of 231/Ctrl mice than those from either tumor free mice or 231/Rab27a KD mice (Fig. 1d), suggesting an elevation of cholesterol metabolism in the liver of 231/Ctrl mice mediated by EVs. Next, we examined the expression levels of the enzymes in the cholesterol biosynthesis pathway in both human (231/Ctrl) and murine breast cancer cell xenograft (4T1/Ctrl) mouse models (Fig. 1e). In the 4T1 mouse model, we also xenografted 4T1 cells with Rab27a gene knockout (4T1/Rab27a KO) cells, which displayed impaired exosome secretion capability when compared to wild type 4T1 cells (Supplementary Fig. 1c–e). Both the RNA and protein abundance of HMGCR, FDFT1, LSS and DHCR7 were highly induced while INSIG1 and INSIG2, negative regulators of HMGCR26, were significantly suppressed in liver tissues from 231/Ctrl mice than its relative control groups (Fig. 1f, g). Accordingly, reduced expression of INSIG1 and INSIG2 but elevated HMGCR levels were observed in immunohistochemistry-stained liver tissues from 231/Ctrl mice (Fig. 1h). A similar pattern was detected in the 4T1 mouse model (Supplementary Fig. 1f, g), suggesting the interorgan communication between the primary tumor and liver in vivo.

a Representative images for GFP signals in liver from mice xenografted MDA-MB-231/Lck-GFP cells (231/Ctrl) or MDA-MB-231/Rab27a KD/Lck-GFP cells (231/Rab27a KD) and tumor free mice. b Liver tissue collected from 231/Ctrl mice and 231/Rab27a KD mice for 50 days and tumor free mice were subjected to RNA-seq and GSEA, showing enrichment of genes related to indicated pathways (Permutation test, n = 3 mice per group). c Total cholesterol (TC) in liver and serum in indicated group (one-way ANOVA, n = 7 mice per group). d Liver sections were stained with oil red O. Scale bars, 50 μm. e Schematic of cholesterol synthesis pathway. f The qRT-PCR showing relative mRNA levels in the liver in indicated group (one-way ANOVA, n = 6 mice per group). g Western blots showing proteins related to cholesterol synthesis in the liver in indicated group. h IHC of indicated protein in liver sections. Representative images are shown. Scale bars, 100 μm. i GFP signals in liver from mice receiving tail vein injections of Lck-GFP-labeled EVs. j Liver tissue collected from mice that had received 5 weeks of i.v. injections of EVs or control mice were subjected to RNA-seq and GSEA, showing enrichment of genes set related to cholesterol biosynthesis pathway (Permutation test, n = 3 mice per group). k Total cholesterol (TC) in liver in indicated group (one-way ANOVA, n = 5 mice per group). l Left: Liver sections were stained with oil red O in NSG mice receiving tail vein injections of EVs. Scale bars, 50 μm. Right: Quantification of the lipid droplets of the liver (one-way ANOVA, n = 5 biological replicates). m Quantification of gene expression of several markers in cholesterol synthesis pathway using qRT-PCR in NSG mice receiving indicated EVs (one-way ANOVA, n = 6 mice per group). n Immunoblot analysis of liver sample collected from mice that had received MCF-10A or MDA-MB-231-derived EVs for 5 weeks. o Immunoblot analysis of proteins related to cholesterol synthesis in primary hepatocytes treated with indicated EVs, with PBS as control. Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
To further verify whether EV resulted in this phenomenon, the GFP-labeled EVs (normal human breast epithelial cell MCF-10A derived EV and MDA-MB-231 EV) were intravenously (i.v.) injected into female NOD/SCID/IL2Rγ-null (NSG) mice semiweekly-for-5 weeks, with liver and lungs collected for downstream bioluminescence imaging (Fig. 1i). We found there were sufficient GFP signals captured in both liver and lung, further confirming that cancer-derived EV could be taken up in vivo (Supplementary Fig. 1h, i). GSEA analysis also enriched the cholesterol biosynthesis pathway in the liver of mice receiving MDA-MB-231 EV compared to the control groups (Fig. 1j). We next performed ELISA quantification and oil Red O staining for the liver from EV-injected mice. We found MDA-MB-231 EV-educated liver exhibited more lipid accumulation than liver from either MCF-10A EV or PBS-injected mice (Fig. 1k and 1l). Similarly, this effect from cancer-derived EV on lipid accumulation was also detected in BALB/c mice receiving 4T1 EV (Supplementary Fig. 1j). Mechanistically, both RNA and protein abundance of cholesterol biosynthesis-related enzymes were upregulated in MDA-MB-231 EV-educated liver from both human (Fig. 1m, n) and murine BC mouse models (Supplementary Fig. 1k, l). Moreover, primary hepatocyte cells showed sufficient uptake of GFP-labeled EVs in vitro (Supplementary Fig. 1m), and MDA-MB-231 EV reduced liver INSIG1 and INSIG2 expression, along with remarkable induction of HMGCR, FDFT1, LSS and DHCR24 levels (Fig. 1o and Supplementary Fig. 1n). Collectively, these results demonstrated breast cancer-derived EVs enhanced cholesterol biosynthesis both in vitro and in vivo.
Circulating miR-9-5p induced cholesterol biosynthesis by targeting INSIG1 and INSIG2
To further explore the underlying mechanism of how cancer EVs regulated cholesterol biosynthesis, we thoroughly searched the potential miRNA pools predicted targeting both INSIG1 and INSIG2, and compared with miRNAs previously reported with higher expression in MDA-MB-231 EV compared to MCF-10A EV13 (see Supplementary Table 1), we found only miR-9-5p co-existed in both databases (Fig. 2a). There was fivefold upregulation of miR-9-5p level in BC serum than in non-cancer patients and threefold enrichment in MDA-MB-231 EV in comparison with MCF-10A EV (Fig. 2b and Supplementary Fig. 2a). Moreover, there were ~40-fold and ~10-fold of miR-9-5p higher expression levels in liver and serum, respectively, from 231/Ctrl mice than the relative control groups (Fig. 2c). In the liver of mice receiving MDA-MB-231 EV, there was a fourfold miR-9-5p higher abundance than those mice receiving either PBS or MCF-10A EV (Fig. 2d). Further in situ hybridization staining evidenced more miR-9-5p expression in the liver from 231/Ctrl mice (Fig. 2e). To examine the biological role of miR-9-5p in EVs, we thereby constructed miR-9-5p overexpressed MCF-10A cells (MCF-10A/miR-9) and demonstrated 15-fold and sixfold of miR-9-5p expression higher within intracellular and EV fraction from MCF-10A/miR-9-5p cells than MCF-10A cells (Supplementary Fig. 2b). Intriguingly, interrogation of sequences near the 3’UTR of human and mouse INSIG genes identified there was each miR-9-5p binding site for human Insig1 and Insig2 while two miR-9-5p binding sites for mouse insig1 and insig2, respectively (Fig. 2f). Luciferase reporter assays comparing miR-9-5p responsiveness of wild-type and miR-9-site-mutated constructs confirmed direct targeting of INSIGs by miR-9-5p through the upstream site in both human and mice (Fig. 2g). Consistently, remarkable reductions of INSIG1 or INSIG2 levels were observed in intracellular MCF-10A/miR-9-5p cells versus MCF-10A cells (Supplementary Fig. 2c), further proving miR-9-5p could target both INSIG1 and INSIG2.

a The enriched miRNAs from MDA-MB-231 EVs versus MCF-10A EVs overlapping with miRNA pools targeting both Insig1 and Insig2 from Targetscan database in cholesterol pathway. b Left: qRT-PCR-determined miR-9-5p levels in an equal amount of EVs derived from serum of normal (n = 6) and breast cancer patients (n = 12) (unpaired two-tailed t test). Right: The RNA quality of serum EVs was demonstrated. c RT-qPCR-determined miR-9-5p levels in liver (n = 5) and serum (n = 5) in indicated tumor-bearing or tumor-free mice (one-way ANOVA). d RT-qPCR-determined miR-9-5p levels in liver from mice receiving tail vein injections of indicated EVs (one-way ANOVA, n = 6 mice per group). e In situ hybridization for miR-9-5p in liver of mice from tumor free, 231/Ctrl and 231/Rab27a KD. Scale bar, 50 μm. f Predicted miR-9-5p binding sites in human and mouse Insigs genes (Insig1 and Inisg2, respectively). The corresponding sequences in WT and mutated reporters are shown. g Luciferase reporter assay showing responsiveness of the reporters to transfected human and mouse Insig1 or Insig2 in MCF-10A and MCF-10A/miR-9 cells (unpaired two-tailed t test, n = 3 independent batches). h Immunoblot analysis of liver sample collected from indicated mice received EVs injection. i, j Total cholesterol (TC) and triglycerides (TG) level in liver and serum in indicated mice received EVs (unpaired two-tailed t test, n = 6 mice per group). k Quantification of gene expression of HL7702 cell after EVs/PBS treatment for 48 h using qRT-PCR (one-way ANOVA, n = 3 independent batches). l Immunoblot analysis of HL7702 cell after EVs/PBS treatment for 48 h. m Immunoblot analysis of liver sample collected from indicated tumor bearing mice. n According to the gates in phasor plot, LD region was segmented in livers of indicated groups. Scale bars, 20 μm. o Total cholesterol (TC) and triglycerides (TG) level in liver (n = 6) and serum (n = 5) in indicated tumor bearing mice (unpaired two-tailed t test). p Liver sections were stained with oil red O in indicated groups. Scale bars, 50 μm. Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
To investigate the in vivo effect of circulating miR-9-5p on cholesterol metabolism in the liver, we performed miR-9-5p EV injection through tail vein after a series of characterizations. Electron microscopy demonstrated MCF-10A EV, MDA-MB-231 EV and MCF-10A/miR-9-5p EV with classic exosome morphology and under 200 nm diameter (Supplementary Fig. 2d). Further nanoflow cytometry showed MCF-10A/miR-9 EV with GFP and CD63 expression on the EV membrane (Supplementary Fig. 2e). Additional density ultracentrifugation confirmed the classical exosome distribution of MCF-10A/miR-9 EV (Supplementary Fig. 2f, g). Mice receiving MCF-10A/miR-9 EVs and MDA-MB-231 EVs (EVs loading high amount of miR-9-5p) showed more abundance of HMGCR but lower INSIG1 and INSIG2 expression in the liver (Fig. 2h). Similarly, there was increased TC and total glyceride (TG) in both liver and sera of mice receiving high miR-9-5p carrying EVs (Fig. 2i, j). Oil Red O staining and quantification further supported more lipids accumulation in the liver of mice receiving high miR-9-5p carrying EVs (Supplementary Fig. 2h). Suppressed INSIG1 and INSIG2 expression were observed in the liver from mice receiving MCF-10A/miR-9 EVs (Supplementary Fig. 2i). Moreover, in vitro cultured primary hepatocytes incubated with MDA-MB-231 EVs or MCF-10A/miR-9 EVs also showed increased HMGCR and DHCR24 expression but lower INSIG1 and INSIG2 levels than PBS or MCF-10A EV treated cells (Fig. 2k, l). To further confirm the effect of miR-9-5p on the regulation of cholesterol synthesis, we knocked out 2 of 3 copies of the miR-9-5p gene in MDA-MB-231 cells (231/miR-9 KO) and 4T1 cell (4T1/miR-9 KO) by employing CRISPR-Cas9 technology (Supplementary Fig. 2j). A series of validation confirmed ~80-90% reduced miR-9-5p abundance in intracellular and EV fractions of 231/miR-9 KO cells and 4T1/miR-9 KO cells (Supplementary Fig. 2k, l). We next xenografted both 231/miR-9 KO cells and 4T1/miR-9 KO cells into mice to validate cholesterol related gene expression in 231/miR-9 KO mice and 4T1/miR-9 KO mice, respectively. There was less HMGCR expression in the liver of the 231/miR-9 KO group compared with 231/Ctrl mice (Fig. 2m). Raman imaging also demonstrated lipids were highly enriched in the liver of 231/Ctrl mice rather than either 231/miR-9 KO or 231/Rab27a KD mice (Fig. 2n and Supplementary Fig. 2m). Consistently, mice xenografted with 231/miR-9 KO cells showed decreased total cholesterol (TC) concentration in both liver and serum, along with decreased TG abundance in liver rather than sera (Fig. 2o). Indeed, oil red O (ORO) staining further strengthened the lipid accumulation was blocked in the liver from 231/miR-9 KO mice when compared to 231/Ctrl mice (Fig. 2p). However, there was no noticeable change in body weight detected between 231/Ctrl mice and 231/miR-9 KO mice (Supplementary Fig. 2n). In summary, our results demonstrated BC-derived miR-9-5p induced HMGCR expression to promote cholesterol synthesis by targeting both INSIG1 and INSIG2 in liver tissue.
Circulating miR-9-5p promoted cholesterol to 25-HC conversion by targeting ATF3
Cholesterol is converted into bile acids in the liver through oxysterols25. GSEA enrichment analysis also highlighted the upregulation of oxysterol derived from cholesterol pathway in the liver from mice receiving MDA-MB-231 EV in comparison with MCF-10A EV (Supplementary Fig. 3a). Intriguingly, the same pathway was highly enriched in the liver from 231/Ctrl mice when compared to either 231/miR-9 KO or 231/Rab27a KD mice (Fig. 3a and Supplementary Fig. 3b), suggesting circulating miR-9-5p involved in oxysterol production from cholesterol. Considering cholesterol could be converted into several kinds of oxysterols including 25-hydroxycholesterol (25-HC), 22(R)-HC, 24S-HC, 27-HC, 7β-HC, 7α-HC, and 4β-HC (Fig. 3b), we performed targeted LC-MS/MS for the liver of mice from both tumor xenograft model and EV injected mice model. We found 25-HC was the only oxysterol enriched in the liver from both 231/Ctrl mice and high miR-9-5p EV-educated mice than their respective control groups (Fig. 3c, d). Elevated 25-HC abundance was also detected in lung from 231/Ctrl mice than 231/miR-9 KO mice (Supplementary Fig. 3c). Subsequent qRT-PCR quantification further validated Ch25h, an enzyme converting cholesterol into 25-HC, was highly induced in the liver of 231/Ctrl mice and mice receiving high miR-9-5p EV than their corresponding control groups in both NSG and BALB/c mice (Figs. 3e, f, Supplementary Fig. 3d). Additionally, we also measured other enzymes catalyzing oxysterol conversion, while no significant changes for either Cyp11a1 or Cyp46a1 gene expressions between liver from corresponding mouse models were detected (Supplementary Fig. 3e, f). These results demonstrated cholesterol was mainly converted into 25-HC in liver under the context of tumor.

a Livers collected from mice xenografted with MDA-MB-231cells and 231/miR-9 KO cells were subjected to RNA-seq and GSEA, showing enrichment of genes related to oxysterol derived from cholesterol pathway (Permutation test, n = 3 mice per group). b Diagram of cholesterol conversion to oxysterol. c LC-MS/MS analysis of oxysterol derived from cholesterol in liver in indicated xenografted NSG mice (one-way ANOVA, n = 5 mice per group). d LC-MS/MS analysis of oxysterol derived from cholesterol in liver in indicated mice receiving tail vein injections of EVs (one-way ANOVA, n = 5 mice per group). e Quantification of gene expression in liver in indicated xenografted NSG mice by qRT-PCR (one-way ANOVA, n = 6 mice per group). f Quantification of gene expression in liver in indicated NSG mice receiving tail vein injections of EVs by qRT-PCR (one-way ANOVA, n = 6 mice per group). g Predicted miR-9-5p binding sites in human and mouse ATF3 genes. The corresponding sequences in WT and mutated reporters are shown. h Responsiveness of wild-type and mutant ATF3 3’UTR reporters to miR-9-5p in MCF-10A or MCF-10A/miR-9 cells. Luciferase reporter assay showing responsiveness of the reporters to transfected wild-type and mutant ATF3 3’UTR in MCF-10A and MCF-10A/miR-9 cell (unpaired two-tailed t test, n = 3 independent batches). i Quantification of gene expression in MCF-10A cell transfected with miR-9-5p mimic or control by qRT-PCR (one-way ANOVA, n = 3 independent batches). j Immunoblot analysis of MCF-10A cell transfected with miR-9-5p mimic or control. k Quantification of gene expression of primary hepatocytes after EVs/PBS treatment for 48 h by qRT-PCR (one-way ANOVA, n = 3 independent batches). l Immunoblot analysis of primary hepatocytes after EVs/PBS treatment for 48 h. m Immunoblot analysis of liver in indicated xenografted mice. n IHC of ATF3 and CH25H in liver sections in indicated xenografted mice. Scale bars, 100 μm. o Immunoblot analysis of liver in indicated mice received tail vein injections of EVs. p IHC of ATF3 and CH25H in liver sections in indicated mice received tail vein injections of EVs. Scale bars, 100 μm. Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
To explore the regulation mechanism between miR-9-5p and CH25H, we searched again for all the potential targets for miR-9-5p (see Supplementary Table 2). Both activating transcription factor 1 (ATF1) and ATF2 were predicted by the “Targetscan” database. Considering ATF3 had been shown to suppress CH25H expression in macrophages31, miR-9-5p overexpression significantly suppressed ATF3 expression rather than ATF1 or ATF2 genes (Supplementary Fig. 3g). Interrogation of sequences near the 3’UTR of human and mouse ATF3 genes identified three miR-9-5p binding sites in humans but only one site in mice (Fig. 3g). Luciferase reporter assays comparing miR-9-5p responsiveness of wild-type and miR-9-5p site-mutated constructs confirmed direct targeting of ATF3 by miR-9-5p through the upstream sites in human (Site I) and mice (Fig. 3h). Transfection of primary hepatocytes with miR-9-5p mimic significantly suppressed ATF3 expression rather than ATF1 or ATF2, leading to increasing expression of CH25H (Fig. 3i, j). There was suppressed ATF3 expression but significant upregulation of CH25H in primary hepatocytes incubation with MCF-10A/miR-9 EVs and MDA-MB-231 EVs (high miR-9-5p containing EVs) (Fig. 3k, l). Besides, higher CH25H but lower ATF3 protein abundance was observed in the liver of 231/Ctrl mice (Fig. 3m, n) and mice receiving high miR-9-5p containing EV than their respective control groups (Fig. 3o, p). Consistently, inverse RNA levels of Atf3 were detected in the liver of 231/Ctrl mice and mice receiving high miR-9-5p containing EV than their respective control groups (Supplementary Fig. 3h). Conclusively, the above findings showed BC-derived exosomal miR-9-5p induced CH25H expression to promote cholesterol conversion into 25-HC by targeting ATF3.
Oncometabolite 25-HC promoted BC liver and lung metastasis
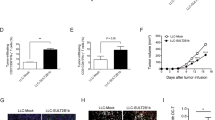
We next investigated the biological effect of miR-9-5p-mediated 25-HC accumulation on tumor progression in vivo. Mice xenografted 231/miR-9 KO cells displayed similar tumor growth rates with 231/Ctrl mice (Fig. 4a), suggesting the energy fuel-independent effect of 25-HC induced by circulating miR-9-5p in vivo. Likewise, miR-9-5p knockout showed a mild impact on the proliferation of MDA-MB-231 cells in vitro (Supplementary Fig. 4a). However, we observed 231/miR-9 KO mice displayed less tumor liver and lung metastasis than 231/Ctrl mice (Fig. 4b, c, Supplementary Fig. 4b). Additionally, there was less Ki67 (proliferation marker of cancer cell) expression in the liver of 231/miR-9 KO mice and 231/Rab27a KD mice, indicating the connection between miR-9-5p secretion and BC metastasis (Fig. 4d, e, Supplementary Fig. 4c). To further investigate the role of 25-HC in vivo, we intraperitoneally injected exogenous 25-HC into 231/Ctrl and 231/miR-9 KO mice (Fig. 4f). We found 25-HC had no obvious effect on tumor growth rate (Supplementary Fig. 4d). Indeed, 25-HC significantly enhanced liver metastasis (Fig. 4g, h), accompanied by more tumor metastatic nodules in the liver (Fig. 4i). The levels of 25-HC in 231/Ctrl mice with 25-HC supplementary was much higher than the other groups (Fig. 4j). Meanwhile, Ki67 expression was further enhanced in 25-HC treated liver tissue (Supplementary Fig. 4e, f). Besides, we found 25-HC promoted both migration and invasion of MDA-MB-231 cells in vitro (Supplementary Fig. 4g, h). Correspondingly, the wound healing assay also indicated 25-HC promoted the migration of cancer cells, simultaneously (Supplementary Fig. 4i, j).

a Tumor volume followed in indicated xenografted mice (two-way ANOVA, n = 5 mice per group). b, c Representative bioluminescence images and quantification of mice inoculated with indicated group (one-way ANOVA, n = 5 mice per group). d, e Representative IHC images showing Ki67 staining and the overall percentage of Ki67+ tumor cells (one-way ANOVA, n = 5 mice per group). Scale bar, 50 μm. f Schematic diagram of 25-HC treated mouse model. 231/Ctrl or 231/miR-9 KO cells injected at day 0. On day 10, mice were treated with vehicle or 25-HC (10 mg/kg) every other day. At day 35, tissue was harvested, imaged and fluorescence quantified. Experimental timeline outlined above quantified data. g, h Representative bioluminescence images and quantification of mice inoculated with indicated groups (one-way ANOVA, n = 5 mice per group). i Quantification of liver nodules (one-way ANOVA, n = 5 mice per group). j 25-HC levels of livers in each group by ELISA kit. (one-way ANOVA, n = 6 mice per group). Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
Ch25h blockage inhibited BC liver and lung metastasis
To further confirm the metastasis-related function of 25-HC in vivo, we knocked down the CH25H expression by injection of AAV8 virus encoding shRNA against CH25H mRNA through the tail vein. There was a significantly decreased CH25H protein abundance as determined by western blotting and IHC in 4T1/shCH25H mice than in 4T1/vehicle mice (Supplementary Fig. 5a). Shrinking tumor volumes were detected in 4T1/shCH25H group (Supplementary Fig. 5b). To confirm the genetic depletion of CH25H, we also employed Ch25h liver-conditional knockout mice (Ch25hL–/–) and validated the decreased plasma 25-HC level in Ch25hL–/– mice than control mice (Fig. 5a). The TC/TG levels and lipids accumulation were also decreased in Ch25hL–/– mice serum and liver (Fig. 5b, c). Positron Emission Tomography-Computed Tomography (PET-CT) is particularly useful for tumor imaging. The results of PET-CT mainly compared the metastases of liver and lung under different treatments, which mainly proved the metastases, just like the previous bioluminescence imaging and Ki67 staining. PET-CT confirmed less metastasis in both the liver and lung of Ch25hL–/– mice (Fig. 5d). Moreover, bioluminescence imaging data showed there were less metastasis sites in both liver and lung from 4T1/shCH25H group (Fig. 5e). We also found reduced number of Ki67-positive cells (Fig. 5f and Supplementary Fig. 5c), along with decreased metastatic nodules in the liver and lung from 231/shCH25H mice (Supplementary Fig. 5d). To further blocked the activity of CH25H, we treated mice with desmosterol, a potent inhibitor of CH25H as a previous report32,33 and observed the same reduced expression of CH25H on metastasis (Fig. 5g). Likewise, bioluminescence imaging data showed there were fewer metastasis sites in both liver and lung from 4T1/desmosterol group (Fig. 5h). Similar effects were further confirmed by Ki67 staining (Fig. 5i). PET-CT also confirmed less metastasis in both the liver and lung of mice treated with desmosterol than the vehicle group (Fig. 5j). The toxicity of desmosterol to hepatocytes was weak (Supplementary Fig. 5e). Compared with the 4T1 control group, mice injected with desmosterol showed no effect on tumor growth rate but successfully maintained body weight especially at a later stage (Supplementary Fig. 5f, g). Besides, desmosterol suppressed both CH25H expression and the proliferation of hepatocytes in vitro (Supplementary Fig. 5h, i). Collectively, 25-HC promoted both liver and lung metastasis while either miR-9-5p knockout or blocking CH25H expression totally abolished the effect of 25-HC on tumor metastasis.

a LC-MS/MS analysis of 25-HC in serum in indicated CH25H liver conditional knock out (Ch25hL–/–) and control (Ch25hfl/fl) mice (unpaired two-tailed t test, n = 6 mice per group). b Total cholesterol (TC) and triglyceride (TG) levels were examined from CH25H liver conditional knock out (Ch25hL–/–) and control (Ch25hfl/fl) mice. (unpaired two-tailed t test, n = 5 mice per group). c Liver sections were stained with oil red O in Ch25h–/– and Ch25hfl/fl mice with E0771 xenograft. Scale bars, 100 μm. d Representative PET/CT imaging with 18F-FDG for accessing liver and lung uptake and quantification in indicated in CH25H liver specific KO and WT mice. e Representative bioluminescence images of metastasis site in liver and lung and quantification of mice inoculated with indicated groups (unpaired two-tailed t test, n = 5 mice per group). f Representative IHC images showing Ki67 staining and the overall percentage of Ki67+ tumor cells in indicated groups (unpaired two-tailed t test, n = 5 per group). Scale bar, 50 μm. g Immunoblot analysis of CH25H in liver in indicated groups (top), IHC and of CH25H in liver sections in indicated groups (bottom). Scale bar, 50 μm. h Representative bioluminescence images of metastasis site in liver and lung and quantification of mice inoculated with indicated groups (unpaired two-tailed t test, n = 5 per group). i Representative IHC images showing Ki67 staining and the overall percentage of Ki67+ tumor cells of liver and lung from indicated groups (unpaired two-tailed t test, n = 5 per group). Scale bar, 50 μm. j Representative PET/CT imaging with 18F-FDG for accessing liver and lung uptake and quantification in indicated mice. Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
25-HC induced Kupffer cell polarization into M2 type by elevating MAFK level to promote liver metastasis
Emerging evidence highlights the fundamental role of immune determinants in the pre-metastatic niche formation34, and our gene enrichment analysis for transcriptome profiling demonstrated that both “macrophage M1 versus M2 down” and “tolerant macrophage up” pathways were upregulated in 25-HC treated macrophages when compared with Vehicle treatment (Fig. 6a). To verify whether liver macrophage-mediated tumor metastasis in the context of the dysregulated cholesterol metabolism, we injected mice with clodronate liposomes to deplete macrophages and accordingly, the macrophage population was largely diminished upon clodronate treatment (4T1/clodronate) when compared to the control treatment (4T1/vehicle) in the liver of tumor bearing mice (Fig. 6b). Indeed, we detected lower F4/80 abundance in both lung and liver tissue upon clodronate treatment (Fig. 6c). Intriguingly, both bioluminescence imaging and immunohistology data demonstrated sharp decrease of tumor metastasis, as well as the reduction of Ki67 in both liver and lung tissue from 4T1/clodronate mice when compared to 4T1/vehicle mice (Fig. 6d, e). Considering Kupffer cells as the major immune cell type in the liver, we subsequently determined whether 25-HC induced liver metastasis by regulation of macrophage polarization. Then, we isolated Kupffer cells from the liver and performed flow cytometry to analyze the polarization signature. We found there was much lower CD163 abundance in Kupffer cells residing in the liver of 4T1/miR-9 KO mice rather than 4T1/Ctrl mice, while 25-HC treatment significantly induced CD163 level, along with the elevation of M2/M1 ratio, indicating M2 macrophage polarization attributed to 25-HC treatment in Kupffer cells (Fig. 6f, g). Beyond, 25-HC treatment also induced other M2 genes including Arg1 and Mrc1 expression in both lung and liver tissues of 231/miR-9-5p KO mice, suggesting the contribution of 25-HC on genetic induction of M2 polarization of macrophages (Fig. 6h and Supplementary Fig. 6a).

a RAW264.7 cells treated with 25-HC and vehicle were subjected to RNA-seq and GSEA, showing enrichment of genes set related to macrophage pathway (Permutation test, n = 3 independent batches per group). b Quantification of purified Kupffer cells with CD11b+ and F4/80+ expression by flow cytometry in indicated groups. c IHC of F4/80 in liver and lung sections in indicated groups. Scale bar, 50 μm. d Representative bioluminescence images of liver and lung metastasis sites in indicated groups. e IHC of Ki67 in liver and lung sections in indicated groups. Scale bar, 50 μm. f Purified Kupffer cells were selected with TIM4+, I-A/I-Eint expression and stained and further analyzed for CD163 and CD11c expression. Representative flow cytometry results were shown. g Quantification of the M2/M1 ratio in flow cytometry results (one-way ANOVA, n = 3 independent batches). h RNA level in liver tissue of mice from indicated groups by qRT-PCR (unpaired two-tailed t test, n = 5 mice per group). i Heatmap showing the relative levels of selected genes related to tolerant macrophage pathway based on the RNA-seq data. j Relative mRNA level of Mafk in liver from indicated mice (unpaired two-tailed t test, n = 6 mice per group) (top); immunoblot analysis of liver in indicated mice (bottom). k The qRT-PCR of Mafk in the RAW264.7 cells after 25-HC treatment with a dose dependent for 48 h (unpaired two-tailed t test, n = 3 independent batches) (top); immunoblot analysis of Raw264.7 cells after 25-HC treatment with a dose dependent for 48 h (bottom). l Different expression and quantification of various chemokines in media derived from 25-HC treated or control-RAW264.7 cells was detected by a mouse chemokine array kit (n = 2 independent batches). Red boxes designate the chemokines whose expression was altered by 25-HC treatment. m TGF-β and IL-10 secreted from BMDM cells treated with vehicle and 25-HC were detected by ELISA kit (unpaired two-tailed t test, n = 6 independent batches). Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
Mechanistically, although “Cistrome Data Browser” predicted MAFK, FOXA1 and RAD21 to activate the transcription of both Arg1 and Mrc1 genes (see Supplementary Table 3), only MAFK was screened out from the enriched tolerant macrophage pathway (Fig. 6i). We found both RNA and protein expression of MAFK were induced by supplementary 25-HC in the liver of 231/miR-9 KO mice (Fig. 6j and Supplementary Fig. 6b). Moreover, MAFK was induced by 25-HC with a dose-dependent manner (Fig. 6k). We detected significantly reduced MAFK protein expression when we knocked down Mafk expression with small interfering RNAs in BMDM cells (Supplementary Fig. 6c, d). Accordingly, Arg1 or Mrc1 were remarkably inhibited due to the MAFK reduction (Supplementary Fig. 6e). M2 macrophages produced growth factors, extracellular matrix-remodeling molecules, and cytokines to regulate cancer migration and metastasis35. Indeed, when we treated RAW 264.7 cells with 25-HC and analyzed the levels of chemokines using a mouse chemokine screening test kit and observed a significant elevation of C-X-C motif chemokine ligand 2 (CXCL2), CXCL10 and CXCL16 secretion into the medium (Fig. 6l). 25-HC also induced the secretion of anti-inflammatory cytokines IL-10 and TGF-β (Fig. 6m). Of note, we also detected the induction of M2 polarization in alveolar macrophages in tumor-bearing mice (231/Ctrl) when compared to tumor free mice (Supplementary Fig. 6f). However, we failed to detect any alteration of key enzymes of cholesterol metabolism in lung tissue, suggesting the possibility of uptake of 25-HC from serum by lung tissue from tumor bearing mice (Supplementary Fig. 6g, h). Collectively, all the above results showed that 25-HC induced the polarization of macrophage into M2 type to promote both lung and liver metastasis.
Exogenous expression of INSIGs and ATF3 blocked miR-9-induced tumor metastasis
We next overexpressed the three targets of miR-9-5p individually or with combination mode following transduction with adeno-associated virus 8 (AAV8) expressing mouse INSIG1, INSIG2 and ATF3 after tumor implantation, with AAV-GFP serving as a control upon additional verification in vitro (Supplementary Fig. 7a). As we replaced the GFP coding sequences with the ATF3 or INSIG1/2 coding sequences on the backbone of AAV plasmid, the green fluorescence signals were hardly detectable in AAV-ATF3 injected mice (Fig. 7a). GFP signals were mainly detected in the liver but with little signal in lung and kidneys from AAV-GFP-injected mice (Supplementary Fig. 7b). Both RNA and protein levels of INSIG1 and INSIG2 were highly expressed in the liver (Fig. 7b, c), accompanied by a significant reduction of TC and TG levels in the mice liver with exogenous expression of INSIG1 and INSIG2 (Fig. 7d and Supplementary Fig. 7c).

a GFP signals in liver and lung from mice receiving tail vein injections of AAV-GFP, AAV-ATF3 and AAV-INSIG1 + INSIG2 virus. Scale bar, 100 μm. b Quantification of gene expression in liver from mice receiving tail vein injections of AAV-GFP, AAV-INSIG1 and AAV-INSIG2 virus. (unpaired two-tailed t test, n = 6 mice per group). c Western blots showing the expression of key proteins related to cholesterol synthesis in liver from mice receiving tail vein injections of AAV-GFP, AAV-INSIG1 and AAV-INSIG2 virus. d Total cholesterol (TC) and triglycerides (TG) in liver in indicated mice (unpaired two-tailed t test, n = 5 mice per group). e Western blots showing protein expression in liver from mice receiving tail vein injections of AAV-GFP and AAV-ATF3 virus. f Representative IHC images showing CH25H staining. Scale bar, 50 μm. g LC-MS/MS analysis of 25-HC level in liver in indicated groups (unpaired two-tailed t test, n = 5 mice for per group). h Body weight was monitored (two-way ANOVA, n = 6 mice per group). i, j Representative bioluminescence images and quantification of mice inoculated with indicated groups (one-way ANOVA, n = 5 mice per group). k Quantification of liver and lung metastatic nodules (unpaired two-tailed t test, n = 6 mice per group). l RNA level of Mki67 in liver and lung in indicated mice (unpaired two-tailed t test, n = 6 mice per group). m Representative IHC images showing Ki67 staining and the overall percentage of Ki67+ tumor cells (one-way ANOVA, n = 5 per group). Scale bar, 50 μm. n Western blots showing proteins in liver from mice receiving tail vein injections of AAV-GFP and AAV-ATF3 virus. o RNA level of M1 and M2 marker gene in liver and lung in indicated mice (unpaired two-tailed t test, n = 6 mice per group). p Purified Kupffer cells were selected with TIM4+, I-A/I-Eint expression and stained and further analyzed for CD163 and CD11c expression. Representative flow cytometry results were shown. q Quantification of the M2/M1 ratio in flow cytometry results (one-way ANOVA, n = 3 independent batches). Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
Meanwhile, AAV-ATF3 restoration effectively inhibited CH25H in the liver from tumor-bearing mice (Supplementary Fig. 7d). Exogenous expression of ATF3 suppressed the protein expression of CH25H (Fig. 7e, f), confirming the central regulatory role of the ATF3/CH25H axis in cholesterol homeostasis. However, there was not too much change for TC and TG concentration in serum and liver under ATF3 overexpression except ~1.5-fold increase of TG accumulation in the liver (Supplementary Fig. 7e, f). 25-HC level was also decreased along with ATF3 overexpression (Fig. 7g). Collectively, we measured the body weight (BW) from mice with INSIG1, INSIG2 and ATF3 overexpression, and observed restored BW but no altered effect on tumor growth rate in AAV-INSIG1 + AAV-INSIG2 or AAV-ATF3 mice than AAV-GFP control mice (Fig. 7h and Supplementary Fig. 7g). Those results further indicated the effect of miR-9-5p mildly contributed to primary tumor growth. Bioluminescence imaging data showed both liver and lung metastasis were blocked with exogenous those tri-targets (Fig. 7i, j). Consistently, there were fewer metastatic sites in both organs from mice treated with the AAV-ATF3 virus (Fig. 7k). Less Ki67 positive cells with high Ki67 expression were detected in both liver and lung from AAV-INSIG1 + AAV-INSIG2 and AAV-ATF3 mice (Fig. 7l, m). Moreover, ATF3 overexpression successfully reduced MAFK abundance in the liver of mice receiving AAV-ATF3 virus (Fig. 7n). Additionally, decreased expression of M2 markers including Arg1, Mrc1 and CD163 were detected in the livers of those mice as well (Fig. 7o, p), along with a reduced M2/M1 ratio (Fig. 7q). Either ATF3 overexpression or INSIG1 or INSIG2 overexpression showed no interference with other targets’ expression (Supplementary Fig. 7h, i). Altogether, by employing a gain-of-function approach to express the three targets exogenously, we demonstrate a significantly restored regulation of cholesterol homeostasis mediated by miR-9-5p.
Silencing of miR-9-5p attenuated 25-HC-mediated tumor metastasis
To verify to what degree the primary tumor regulated the metastasis through circulating miR-9-5p, we injected miR-9-5p antagomir into the tail vein of both human (MDA-MB-231) and murine (4T1) cells xenografted mice (Fig. 8a). The miR-9-5p antagomir treatment remarkably decreased in vivo miR-9-5p levels in the serum, tumor, liver and lung tissue of mice (Fig. 8b). Besides, we failed to detect any significant alterations of either tumor size among miR-9-5p antagomir and the control group or miR-9 KO and Rab27a KD, which further confirmed that miR-9-5p displayed mere regulatory effect on tumor growth (Supplementary Fig. 8a). However, PET-CT captured significantly reduced metastasis niches in both liver and lung from the mice receiving antagomir miR-9-5p treatment (Fig. 8c). Moreover, bioluminescence imaging data further strengthened that miR-9-5p antagomir blocked the metastasis in both BC xenograft models (Fig. 8d, e, and Supplementary Fig. 8b, c). Mechanistically, miR-9-5p antagomir relieved the inhibitory expression of tri-targets (Insig1, Insig2 and Atf3), and subsequently reduced the RNA levels of Hmgcr and Ch25h (Fig. 8f). Besides, miR-9-5p antagomir treatment resulted in a reduction of the M2 marker genes abundance but an elevation of M1 marker genes level (Fig. 8g). Simultaneously, miR-9-5p antagomir also restored the expression of ATF3 and blocked the expression of CH25H and HMGCR (Fig. 8h). The TC and TG levels in liver were also decreased by miR-9-5p antagomir (Supplementary Fig. 8d). Collectively, silencing of miR-9-5p attenuated 25-HC-mediated tumor metastasis by switching macrophage polarization, suggesting the potential therapeutic applications for targeting both circulating miR-9-5p and 25-HC biosynthesis.

a Schematic diagram of antagomir treated mouse model. 231/Luc cells injected at day 0 in NSG mice. On day 14, tumor was detected. At day 28, mice were treated with antagomir (10 nmol) every 3 days untiled euthanized. 4T1/Luc cells injected at day 0 in BALB/c mice. On day 8, tumor was detected. At day 25, mice were treated with antagomir (10 nmol) every 3 days untiled euthanized, during which time the tumor was removed on day 28. b RT-qPCR-determined miR-9-5p levels in serum, tumor, liver and lung in 231/Luc cells xenografted mice treated with indicated antagomir and with 231/Ctrl, 231/Rab27a KD and 231/miR-9 KO mice (one-way ANOVA, n = 5 mice per group). c Representative PET/CT imaging with 18F-FDG for accessing liver and lung uptake and quantification in 4T1/Luc cells xenografted mice treated with indicated antagomir. d Representative bioluminescence images of metastasis site in liver and lung of xenografted mice treated with indicated antagomir and with 231/Ctrl, 231/Rab27a KD and 231/miR-9 KO mice. e Quantification of liver and lung metastasis of xenografted mice treated with indicated antagomir and with 231/Ctrl, 231/Rab27a KD and 231/miR-9 KO mice (one-way ANOVA, n = 5 mice per group). f Quantification of gene expression in liver from xenografted mice treated with indicated antagomir and with 231/Ctrl, 231/Rab27a KD and 231/miR-9 KO mice (one-way ANOVA, n = 5 mice per group). g Quantification of gene expression in liver from xenografted mice (one-way ANOVA, n = 5 mice per group). h Immunoblotting analysis of liver sample collected from xenografted mice treated with indicated antagomir and with 231/Ctrl, 231/Rab27a KD and 231/miR-9 KO mice. Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
Circulating miR-9-5p was correlated with BC patients with metastasis
To investigate whether the mechanism of miR-9-5p-mediated cholesterol metabolism and tumor metastasis widely exists in breast cancer, we examined the miR-9-5p expression in BC cell lines under different subtypes. We found a relatively higher level of miR-9-5p detected in intracellular MCF-7, BT474, SK-BR-3, 4T1 and E0771 cells and their derived EV fraction than the level of intracellular and EV fraction from MCF-10A cells (Fig. 9a, Supplementary Fig. 9a). We also expanded our current mouse model to investigate the role of miR-9-5p in the mice xenografted with distinct TNBC cell line (MDA-MB-468), MDA-MB-231-Lung Metastasis-2 (231-LM2), which displayed higher capability of lung metastasis, as well as ER positive BC cell line (MCF-7). Intriguingly, we observed that miR-9-5p knockout also significantly inhibited the liver and lung metastasis, especially blocking their liver macrophage polarization into M2 type in 468/miR-9 KO mice when compared to 468/Ctrl mice (Supplementary Fig. 9b, c). However, there was no effect on tumor growth due to miR-9-5p knockout (Supplementary Fig. 9d). Similar pattern was detected in mice xenografted with 231-LM2/miR-9 KO tumor when compared to their wild type tumors (Supplementary Fig. 9e–g) as well as MCF-7/miR-9 KO tumor versus MCF-7 tumor (Supplementary Fig. 9h, i). All the above data showed miR-9-5p was generally correlated with liver metastasis of mice xenografted with distinct BC types. Beyond, more circulating miR-9-5p levels were also detected in mice xenografted with lung cancer (A549), hepatocarcinoma (Hep3B) and colon cancer (SW480) models (Fig. 9b).

a RT-qPCR-determined miR-9-5p levels in an equal amount of EVs derived from indicated cells (normalized to miR-16) (unpaired two-tailed t test, n = 3 independent batches). b RT-qPCR-determined miR-9-5p levels of EVs in serum from indicated xenografted mice. (one-way ANOVA, n = 3 independent batches). c Representative images of miRNA in situ hybridization (miRNA-ISH) in liver metastases from breast cancer patients. Scale bar, 50 μm. d RT-qPCR-determined miR-9-5p levels in serum from normal or BC patient with or without metastasis (unpaired two-tailed t test, n = 5). e Expression levels of miR-9-5p in BRCA tumor samples from the TCGA database. Statistical significance was assessed using two-tailed Mann–Whitney test (left, n = 1102 for Tumor and n = 104 for Normal) and paired two-tailed Student’s t test (right, n = 103). f Expression levels of miR‑9-5p in BRCA tumor samples from the GEO database. Box plots show minima, maxima, median bounds of box and quartiles (one-way ANOVA for left, n = 59 for Normal, n = 169 for Tumor, n = 54 for Metastasis; paired two-tailed t test for right, n = 59). g Expression levels of miR-9-5p in different stage of BRCA tumor samples. Box plots show minima, maxima, median bounds of box and quartiles (one-way ANOVA, n = 104 for normal, n = 182 for stage I, n = 609 for stage II, n = 244 for stage III, n = 20 for stage IV). h IHC analysis of HMGCR, CH25H, INSIG1, INSIG2 and ATF3 expression in liver metastases from breast cancer and non-BC individuals. Scale bar, 100 μm. i LC-MS/MS analysis of oxysterol derived from cholesterol in serum from normal and BC patients (unpaired two-tailed t test, n = 15 per group). j Correlation analysis of MRC1 and Ki67 expression IHC from BC patients metastatic liver. The two-tailed Pearson’s r was indicated (n = 7). k Schematic summary of this study. Data are presented as mean ± s.d. Source data and exact P value are provided as a Source data file.
In situ hybridization (ISH) demonstrated higher miR-9-5p levels in the liver tissue of BC patients with liver metastasis (Fig. 9c). BC patients with metastasis also have higher serum miR-9-5p levels than healthy individuals or patients without metastasis (Fig. 9d). Interestingly, according to the TCGA database, miR-9-5p was highly expressed in primary BC tumors than in adjacent normal tissues (Fig. 9e). Of note, miR-9-5p was highly expressed in patients with both triple-negative BC (TNBC) tumors (GSE45498) and lymph node metastatic lesion tissues (Fig. 9f). Moreover, higher miR-9-5p level was detected in patients of Stage I to Stage IV BC patients (Fig. 9g). We also examined the expression of cholesterol biosynthesis-related enzymes in BC patients with high metastasis (high Ki67 level) or low metastasis (low Ki67 level) and non-BC individuals. IHC demonstrated both HMGCR and CH25H were highly expressed but INSIG1, INSIG2 or ATF3 displayed much lower abundance in BC patients with liver metastasis when compared to non-BC or patients with low liver metastasis (Fig. 9h). Both TC and TG concentrations were significantly elevated in BC patients (Supplementary Fig. 9j). Clinically, significantly elevated plasma 25-HC levels from BC patients than in non-cancer individuals (Fig. 9i). MiR-9-5p was highly expressed in primary BC tumors than in adjacent normal tissues among different types of breast cancer (Supplementary Fig. 9k). With more aggressive liver metastasis, the expression of MRC1 and M2 marker, was also detected with high expression simultaneously (Fig. 9j and Supplementary Fig. 9l). Collectively, our finding reveals the circulating miR-9-5p functions as “tri-brakes” to regulate cholesterol homeostasis and 25-hydroxycholesterol as an oncometabolite to induce BC metastasis by regulating Kupffer cell polarization in the liver (Fig. 9k).
Discussion
Here, we identify the biological function of circulating miR-9-5p encapsulated by EVs and 25-hydroxycholesterol as oncometabolite in liver and lung metastasis of BC. Functioning as “tri-brakes”, miR-9-5p is endowed with three key targets in this study. The first two targets are insulin-induced genes including Insig-1 and Insig-2, widely involved in cholesterol metabolism, lipogenesis, and glucose homeostasis. Following the third target is Atf3, functioning as the repressive transcription factor of CH25H expression. A recent report further supports the fundamental role of the ATF3-CH25H axis in cancer, primarily working on the attenuation of anti-tumor immunity, stimulation of tumor growth, and impeding the efficacy of chimeric antigen receptor (CAR) T cell adoptive therapy36. Therefore, miR-9-5p partially erases the inhibitory regulation of cholesterol biosynthesis and enhances cholesterol to 25-HC conversion by targeting Insig1, Insig2 and Atf3. Additionally, miR-9-5p has also been reported with other targets in the cholesterol metabolism pathway including acyl-coenzyme A: cholesterol acyltransferase (ACAT), a key enzyme that catalyzes the formation of cholesteryl esters (CE) from cholesterol and long-chain fatty acyl-coenzyme A37. Those studies further strengthen the effect of miR-9-5p as a master regulator on cholesterol homeostasis in vivo.
In our study, we demonstrate the effect of 25-HC on the induction of liver Kupffer cells polarization into M2 type, which further facilitates metastatic BC cells to escape from immune surveillance to promote liver metastasis. We found that lung metastasis of breast cancer may be caused by the accumulation of 25-HC synthesized by the liver in the lungs through blood transport, leading to M2 polarization of alveolar macrophages. Indeed, the 25-HC level has been evidenced with a remarkable elevation in lipopolysaccharide (LPS)-induced acute lung inflammation and its sulfated metabolite, 25-hydroxycholesterol-3-sulfate, exerts a strong suppression effect on neutrophil38. More reports show tumor cells induce neutrophils to form neutrophil extracellular traps, thereby supporting lung colonization of metastasis-initiating BC cells39. The recent finding also reveals nicotine promotes BC metastasis by stimulating N2 neutrophils and generating a pre-metastatic niche in the lung40. Besides, lipid-laden lung mesenchymal cells reshape the local immune landscape via metabolic reprogramming of lung-resident fibroblasts to promote BC metastasis41,42.
Identifying secreted regulators in cancer incidence and metastasis determinants provides potential prognostic markers and therapeutic targets for cancer intervention43. Our study reveals the possibility of employing miR-9-5p as a novel blood-based biomarker to identify BC patients with a high risk of developing metastasis to guide preventive screenings and early intervention. Considering CH25H is the key enzyme in our model to regulate tumor metastasis, targeting this expression or blocking its activity might be a powerful strategy for clinical applications. Proteolysis-targeting chimeras (PROTAC) are becoming an emerging therapeutic modality with the potential to tackle disease-causing proteins that have historically been highly challenging to target with conventional small molecules44,45. Therefore, the investigation of targeting CH25H with PROTACs would be a powerful strategy for downstream clinical applications to treat cancer.
Methods
Cells and constructs
Cell lines applied in this study were obtained from American Type Culture Collection (ATCC) and cultured in the recommended medium or as indicated. These include MDA-MB-231 (HTB-26), 4T1 (CRL-2539), SK-BR-3 (HTB-30), MCF-7 (HTB-22), E0771 (CRL-3461), MCF-10A (CRL-10317), BT474 (HTB-20), HEK 293 T (ACS-4500), RAW264.7 (TIB-71). The HL-7702 human hepatocyte cell line was purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences. MDA-MB-468 (HTB-132) and MDA-MB-231-Lung Metastasis-2 (MDA-MB-231-LM2) cell lines were generous gifts from Ke Gong, Wuhan University and Qian Zhao, Shanghai Jiao Tong University. Primary hepatocytes were obtained from mice livers based on the classic two-step collagenase perfusion technique46. Kupffer cells were prepared by two-step liver collagenase digestion and fractionation on a two-step percoll density gradient, as previously described47. All cell lines were cultured in an atmosphere of 5% CO2 incubator at 37 °C. MCF-10A cells were engineered to stably overexpress miR-9-5p (MCF-10A/miR-9) or the empty PLVX-IRES-zsGREEN vector (MCF-10A/vec). MDA-MB-231/Lck-GFP and MCF-10A/Lck-GFP cells were constructed by transfection of an Lck-GFP expression plasmid (Addgene #61099) into MDA-MB-231 and MCF-10A cells followed by selection in G418 and sorting of GFP+ cells by Flow Cytometry. MDA-MB-231/Luciferase and 4T1/Luciferase were constructed by infected MDA-MB-231 and 4T1 cells with packaged plenti-CMV-Puro-Luc virus (Addgene #17477). To construct 231/Rab27a KD cells, short hairpin RNA targeting Rab27a sequence CCAGTGTACTTTACCAATATA (231/Rab27a KD) were designed by BLOCK-iT RNAi Designer (Thermo Fisher Scientific) and the corresponding oligonucleotides were cloned into pLKO.1 vector (Addgene #10878). Cells were transduced by the lentivirus and selected based on puromycin resistance.
To knock out hsa-miR-9-5p gene by employing CRISPR-Cas9 genomic editing system, two single guide RNAs (sgRNAs; DNA sequences ACTCCTACCTGTGCCGACTG and GGGACACGAGTGGAGTTGAG for hsa-miR-9-1; DNA sequences TCATCCCACCTTTAATCATA and AAAAGGAGGAATCTTAAGCG for hsa-miR-9-2) predicted by the sgRNA Designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) were synthesized in DNA form and annealed into double strands, treated with T4 polynucleotide kinase and inserted into the BbsI-digested Lenti-crispr-V2 vector. The two constructs, psPAX2 and pMD2.G were co-transfected into 293 T cells. Virus was harvested at 72 h post transfection and filtered through a 0.45 μm filter. MDA-MB-231, MDA-MB-468, MCF-7 and MDA-MB-231-LM2 cells were infected with collected lentiviruses with the addition of polybrene, selected by puromycin resistance and seeded on 96-well plates. Monoclonal cell was screened by genotyping PCR and confirmed by sequencing. A similar strategy was applied to generate 4T1/miR-9 KO cells by using two sgRNAs (DNA sequences GCTCCGAGCTACCGACTGCA and AGTCCGGAGAAGCCGCTCCC) and 4T1/Rab27a KO cells using sgRNA (DNA sequence AGCGTCCCTGAAGAATGCAG). To construct AAV-mINSIG-1, AAV-mINSIG-2 and AAV-mATF3, the enhanced GFP (eGFP) region in pscAAV-CMV-eGFP plasmid48 was replaced with the DNA fragment containing mouse Insig1, Insig2 and ATF3 cDNAs. The resulting construct pscAAV-CMV-mouse Insig1, pscAAV-CMV-mouse Insig2 and pscAAV-CMV-mouse Atf3 were confirmed by sequencing. AAV serotype 8 was used to achieve robust transgene expression in liver. Viruses were produced and titer was determined by qPCR. For mouse injection, 5 × 1011 genome copy resuspended in 100 μL PBS was used per mouse.
To construct reporters for miR-9-5p-mediated regulation of INSIG1, INSIG2 and ATF3, 3′ UTR of human or mouse INSIG1 (primers 5ʹ- gaaaaaaatttccaatgaagatgtcagca and 5ʹ-gtaaaaaatgaatgcaaaaagaacacaatg for human; primers 5ʹ- gggacggttcttaaagtttcctgg and 5ʹ- gcagcattgtgttcttttgcattct for mouse), INISG2 (primers 5ʹ-aaagcatatggggagctggtattt and 5ʹ- tgcagatgacagctaatatcactttgc for human; primers 5ʹ-gccaaacgtgttactgacacaga and 5ʹ-aggccctctatagctgctcagtca for mouse) and ATF3 (primers 5ʹ-atgcccaccgttaggattcag and 5ʹ- accatgtcttggttccaatatttattattctg for human; primers 5ʹ- cttgataggtggggtgggac and 5ʹ- gttccgatatttattatgttgacaggctag for mouse) gene were cloned by PCR and inserted into the psiCHECK-2 reporter vector (Promega) downstream of the Renilla luciferase (R-Luc) gene. Site-directed mutagenesis was used to mutate the following putative miR-9-5p binding sites with corresponding primer sets: 5ʹ-cagcgtttgatttgagtttgtaaaaatgtaaaa and 5ʹ-tcatatattttctataaactattggcaaaaaaatcc for human INSIG1; 5ʹ-aactttgagggaggttgatggtgcttaa and 5ʹ-gcactgacacaaaactgctagttgg for mouse Insig1 site I; 5ʹ-taactcttgactgagtttgtacaaatggta and 5ʹ-ttgttgtaactgctataactattggcaaaagc for mouse Insig1 site II; 5ʹ- ttggttcgtaattttctgttttgacagg and 5ʹ-gctgtctccagtaacgcaggtttgaaa for human INSIG1; 5ʹ-ttcgtatatactttcccgatttcttcagaag and 5ʹ- gtccctacaacaatgcagacttaaaacc for mouse Insig1 site I; 5ʹ- aatctatgatgctaattgtcactggctact and 5ʹ- cctctgtggcagagagagggcta for mouse Insig1 site II; 5ʹ- ctcagttctcctatcacaggaagaaagcagaaa and 5ʹ-tcctgtgataggagaactgagtttgcaacaga for human ATF3 site I; 5ʹ-ttcaacttctcgtaggttaggactctccactcaa and 5ʹ-tcctaacctacgagaagttgaactttctgctttctt for human ATF3 site II; 5ʹ-ctggaagagctctcaaatattccattttcctttcc and 5ʹ- tggaatatttgagagctcttccagcctagacacaact for human ATF3 site III; 5ʹ- aggaagagatgctaataaccacagccacagcctgt and 5ʹ-tggttattagcatctcttcctgccctgtcactga for mouse Atf3 site III.
EV purification and characterization
EVs were purified by ultracentrifugation of conditioned medium as previously described13,49,50. Briefly, conditioned medium was collected from cells grown in medium containing EV-depleted serum for 48 h, and pre-cleared by centrifugation at 500 × g for 15 min and then at 10,000 × g for 20 min. EVs were pelleted by ultracentrifugation at 110,000 × g for 70 min, and washed in PBS following the same ultracentrifugation conditions. The EV pellet was suspended in PBS and used in various experiments. For cell treatment, 2 μg of EVs (equivalent to those derived from ~5 × 106 producer cells) based on protein measurement by BCA protein assay kit (Thermo Fisher Scientific) was added to 2 × 105 recipient cells in a six-well plate. The characterization of EVs was confirmed by measuring expression of exosome-specific markers TSG101, ALIX and EV-associated protein markers CD63, and CD9 by western blot analysis and nano-flow cytometer (CytoFlex S, Beckman Coulter). In addition, we further examined the characteristics of EVs using iodixanol gradient centrifugation as reported by Kowal et al.51. EVs size distribution and particle concentration were analyzed using Flow NanoAnalyzer U30 (NanoFCM, Xiamen, China) according to the manufacturer’s instructions. Briefly, the instrument was calibrated for particle concentration to 107-109 particles/mL using 250 nm fluorescent silica nanospheres (NanoFCM Inc., QS2503) and for size distribution using Silica Nanospheres Cocktail (NanoFCM Inc., S16M-Exo). EVs were incubated with APC-CD63 antibody at room temperature for 30 min for APC positive detection. The report for size distribution and particle concentration was generated directly from the NF Profession 2.0 software, with the particles from PBS subtracted. Flow cytometer verified the EVs quantity of Rab27a knock out or knock down outcomes in 4T1 and MDA-MB-231 cells. The instrument calibrated for gating EVs with 100 nm and 200 nm Latex Beads (PCS Control Mixed Kit, Beckman Coulter, 6602336). The report for size distribution and particle concentration was generated directly from CytoFLEX 2.4 software.
RNA extraction and quantitative PCR with reverse transcription
Total RNA was harvested from cultured cells or tissues using the TRIzol reagent (Invitrogen, 15596026) followed by cDNA synthesis using the MonScript RTIII Super Mix with dsDNase kit (Monad Biotech, MR05201). A CFX Connect real-time PCR system (Bio-Rad Laboratories) was used to perform quantitative real-time PCR (RT-qPCR) of cDNA samples using MonAmp ChemoHS qPCR Mix (Monad Biotech, MQ00401). The exosomal RNAs were extracted by TRIzol LS reagent (Invitrogen,10296028). EVs resuspended in 1 mL TRIzol LS and then incubated at room temperature for 10 min. 200 μL of chloroform was added, gently mixed, and incubated for 10 min on ice. After incubation, microcentrifuge tubes were centrifuged for 10 min at 12,000 × g at 4 °C. The aqueous phase was transferred to new tubes, an equal volume of isopropanol and 1 μL glycogen (Roche, 10901393001) were added and mixed with incubation at –20 °C overnight. Samples were centrifuged at 12,000 × g for 15 min at 4 °C. Aspirated off liquid and added 500 μL of cold 75% ethanol to wash the pellet. The RNA pellet was air dried for a few minutes at room temperature and subsequently resuspended in 20 μL of RNase-free water. The cellular or exosomal miRNA was reverse transcribed according to the miScript II RT Kit (QIAGEN, 218161) and measured using miScript SYBR Green PCR Kit (QIAGEN, 218073). The primers for miR-9-5p, miR-16 and U6 were purchased from Tiangen (Cat# CD201-0142, CD201-0235 and CD201-0145). All the other primers were seen in Supplementary Table 4.
Western blot analysis
Cells or tissues were homogenized in lysis buffer containing 50 mM Tris (pH 7.4), 500 mM NaCl, 1% NP40, 20% glycerol, 5 mM EDTA and 1 mM PMSF, 1 mM DTT (Roche), supplemented with protease and phosphatase inhibitor cocktails (Roche). Proteins were separated by electrophoresis on a 10% or 12% SDS polyacrylamide gel. Protein detection was performed using the antibodies described in Supplementary Table 5.
Immunohistochemistry (IHC), Immunofluorescence (IF) and in situ hybridization (ISH)
Mouse liver and lung were fixed in 10% neutral buffered formalin and embedded in paraffin for sectioning. Immunohistochemistry (IHC) and immunofluorescence (IF) were performed as previously described50. In situ hybridization (ISH) analysis following previously reported protocol22. Ki67 IHC was used to calculate Ki67+ cells against total cell number from >4 views in each mouse. The antibodies used were listed in Supplementary Table 5. The hsa-miR-9-5p probe with ʹ5’ DIG and 3ʹDIG (5ʹ UCAUACAGCUAGAUAACCAAAGA3’) was purchased from Sangon Biotech (Order number: R11929).
Oil red O staining
Snap frozen liver specimens were sectioned at 6 μm and fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS after being restored to room temperature and then washed twice with PBS. Once completely air-dried at room temperature, cells were further incubated with Oil Red O working solution at room temperature for 1 h. Cells were washed 3 times with PBS before images were acquired for downstream analysis.
Plasmid transfection and siRNA treatment
HEK293T cells were transfected using Lipofectamine 2000 (Life Technologies) with packaging plasmid: psPAX2 (Addgene, #12260) and pMD2.G (Addgene, #12259). Forty-eight hours after transfection, lentiviral supernatant was harvested and filtered through a 0.45-μm filter, and utilized for subsequent infection of MCF-10A, MDA-MB-231 and 4T1 cells (48 h incubation) together with 10 μg/mL polybrene in the medium. Cells were selected based on puromycin selection or sorted by GFP+ cells with FACS. For MAFK knockdown, siRNAs designed for MAFK (siG150120091935 and siG150120091948) and control (siN000000-1-1-5) were purchased from RiboBio (Guangzhou, China).
RNA-seq and gene set enrichment analysis (GSEA)
RNA-seq and data analysis were carried out by Shanghai Majorbio Bio-pharm Technology company. RNA was extracted according the manufacturer’s instructions (Invitrogen) and genomic DNA was removed using DNase I (TaKara). Then RNA quality was determined by 2100 Bioanalyser (Agilent) and quantified using the ND-2000 (NanoDrop Technologies). RNA-seq transcriptome library was prepared following TruSeq RNA sample preparation Kit from Illumina (San Diego, CA) using 1 μg of total RNA. Each library was sequenced on the Illumina HiSeq xten/NovaSeq 6000 platform using paired-end, non–strand-specific sequencing strategy. The raw paired end reads were trimmed and quality controlled by SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) with default parameters. Then clean reads were separately aligned to reference genome with orientation mode using HISAT252 software. The mapped reads of each sample were assembled by StringTie (https://ccb.jhu.edu/software/stringtie/index.shtml? t=example) in a reference-based approach53. To identify DEGs (differential expression genes) between two different samples, the expression level of each transcript was calculated according to the transcripts per million reads (TPM) method. RSEM54 was used to quantify gene abundances. Essentially, differential expression analysis was performed using the DESeq2 /DEGseq55 /EdgeR56 with Q value ≤ 0.05, DEGs with |log2FC | >1 and Q value <= 0.05(DESeq2 or EdgeR) /Q value <= 0.001(DEGseq) were considered to be significantly different expressed genes. For gene set enrichment analysis (GSEA), fragments per kilobase million was uploaded to GSEA 4.2.0 and enrichment of gene sets was interrogated with 1000 random permutations to obtain the normalized enrichment score (NES), nominal P value and false discovery rate (FDR) q value.
Measurement of total cholesterol (TC) and triglycerides (TG)
Mice blood was collected by retro-orbital bleeding. The livers were homogenized and supernatants were collected for lipid extraction. Serum from breast cancer patients and healthy individuals were collected from Renmin Hospital of Wuhan University. Total cholesterol and triglyceride levels were determined by the total cholesterol kit and triglyceride kit according to the manufacturer’s instructions (Kehua Bioengineering).
LC-MS/MS analysis of oxysterols
Analysis of sterols were performed on an Agilent 1290 ultrahigh performance liquid chromatography system connected to an Agilent 6460 triple-quadrupole mass spectrometer (UHPLC-QQQ-MS) equipped with a dual AJS electrospray ionization source (Agilent Technologies, Inc.). Sample preparation was followed a previously reported method57 with some modifications. In brief, coprostanol as an internal standard was added to serum and tissue samples. Saponification was carried out in 1 N ethanolic KOH at 37 °C for 1 h. After addition of distilled water, sterols were extracted twice with n-hexane and evaporated to dryness. The reagent mixture for derivatization consisted of picolinic acid (PA), 2-methyl-6-nitrobenzoic anhydride (MNBA), 4-dimethylaminopyridine (DMAP) with molar ratio 2:2:1 (PA: MNBA: DMAP) in anhydrous pyridine. The freshly prepared reagent mixture was added to the dried serum and tissue extraction, and the reaction mixture was allowed to stand at 60 °C for 10 min. After evaporation to dryness, the residue was redissolved in acetonitrile for UHPLC-QQQ-MS analysis. The samples (2 μL) were individually injected on an UHPLC column (Agilent Zorbax Eclipse plus C18, 2.1 mm × 100 mm, 1.8 μm) with temperature at 40 °C. Ultrapure water containing formic acid (0.1%, v/v) and acetonitrile were used as respective mobile phase A and B with flow rate of 0.6 mL/min. The gradient elution scheme was set as follows: 0 min (80% B), 2–4 min (90% B), 7 min (100% B). Mass spectra were recorded with positive MRM mode (530-146 and 635-146 for oxysterols, 516-146 for coprostanol). MassHunter Workstation software (Agilent Technologies, Inc.) was used for data analysis. All the oxysterols examined were summarized in Supplementary Data 1.
Cell migration and invasion assay
To evaluate the migration and invasion capacity of cells, 24-well plates equipped with cell culture inserts containing 8.0 μm pore size membrane were used. Briefly, cells were suspended in 100 μL of serum-free medium and placed in the upper compartment chambers. The lower chamber was filled with 10% FBS as the chemoattractant. For invasion assay, diluted extracellular matrix gel coated the inserts preincubated for 30 min. In all, 5 × 104 cells incubated for the migration assay and 1 × 105 cells incubated for the invasion assay. At the end of the experiments, cells on the upper surface of the filters were wiped, and migrated cells on the lower surface were fixed in 4% paraformaldehyde and stained by 0.1% crystal violet. For wound healing assay. Briefly, cells were seeded into wells of 6-well plates and cultured for 48 h to reach 95%–100% confluence. The cells were then vertically scratched using a sterile pipette tip. Fresh culture medium was added to the cells. Cell migration (wound/scratch closure) was determined based on the images obtained with inverted optical microscopy every few hours.
Flow cytometry
For liver macrophage analysis, NPCs were prepared by two-step liver collagenase digestion and fractionation on a two-step percoll density gradient, as previously described47. TruStain Fc is specific to the common epitope of CD16/CD32 (Biolegend, Cat# 101319). It is useful for blocking non-specific binding of immunoglobulin to the Fc receptors. Zombie Aqua™ is an amine-reactive fluorescent dye (Biolegend, Cat#423101). The antibodies for surface staining were CD45 (Biolegend, Cat#103145), Ly-6G (Biolegend, Cat#127611), CD3 (Biolegend, Cat# 155611), CD19(Biolegend, Cat# 115526), F4/80 (Biolegend, Cat#123119), CD11b (Biolegend, Cat#101227), CD11c (Biolegend, Cat# 117307), CD163 (Biolegend, Cat#155319), Tim4 (Biolegend, Cat#130021) and I-A/I-E (Biolegend, Cat#107627). Numbers obtained were subsequently represented as the percentage of the highest subsets. Un-stained, single-stained, and the FMO (fluorescence minus one) control were used for setting compensation and gates.
Electron microscopy (EM)
For electron microscopy, EVs were loaded on formvar and carbon-coated copper grids for 1 min. Then the grids were rinsed with 1x PBS before being left for 30 s on 2% (wt/vol) uranyl acetate (Ladd Research Industries, Williston, VT). Excess solution was removed with Whatman 3MM blotting paper, and grids were left to dry for a few minutes before viewing. Grids were examined using a JEOL JEM-1400 Plus transmission electron microscope operating at 80 kV. Images were recorded using a GATAN OneView 4 K digital camera.
Stimulated Raman scattering (SRS) imaging of liver tissue
The liver tissue samples were sliced using a cryostat at 20 μm thickness. Before SRS imaging, the liver tissue samples were washed with PBS solution for 5 min. Subsequently, the liver tissue samples were fixed by 10% neutral formalin (HT501128, Sigma-Aldrich) for 15 min. Finally, the liver tissue samples were washed with PBS solution and immersed in PBS solution for SRS imaging. SRS imaging was performed on a home-built picosecond SRS microscope. An ultrafast laser system with dual output at 80 MHz (picoEmerald, Applied Physics & Electronics) provided both pump (tunable wavelength 700–990 nm) and Stokes beams (fixed wavelength 1031 nm). The Stokes beam was modulated at 20 MHz by an electro-optic modulator. A 60× water objective (numerical aperture 1.2, Olympus) was used to focus the light into the sample. Then, an oil condenser (numerical aperture 1.4, Olympus) was used to collect the forward-going pump and Stokes beams through the specimen. A short-pass filter (1000 nm SP, Thorlabs) was used to block the Stokes beam and transmit only the pump beam onto a large area photodiode (10 × 10 mm) to detect the stimulated Raman loss signal. The powers of the pump and the Stokes beams at the specimen were maintained at ~45 mW and ~45 mW, respectively. The images were acquired at 10 μs pixel dwell time. For hyperspectral SRS (hSRS) imaging of the C-H stretching vibration modes, the pump beam was tuned between 784.4 and 800.0 nm while the Stokes beam was fixed at 1031 nm to obtain 50 image stacks within 3050–2800 cm–1.
In situ LD analysis of hSRS image stack
Phasor analysis was used to segment LDs from the tissue samples based on the SRS spectral profile58. In previous report, the spectrum in the 2750–3050 cm–1 is well fitted as a sum of seven Lorentzian bands59,60. We apply those seven Lorentzian bands and an additional C = C-H stretching vibration band to multi-peak fitting. The Lorentz peak function (y = y0 + (2*A /π) * (w / (4*(x - xc) ^2 + w^2))) were used for multi-peak fitting in OriginPro 2021 according to fitting parameter in Supplementary Table 6. The max height of Lorentz peak in ~3015 cm−1 is used to quantify the unsaturation level.
Animals
Animal experiments in this study were approved by and performed in accordance with the institutional animal care and use committee (IACUC) at the College of Life Science, Wuhan University (WDSKY0202008). All mice used in this study were purchased from Shanghai Model Organisms Center, Inc. (NSG mice, Cat.no. NM-NSG-001), Nanjing GemPharmatech (BALB/c-Nude mice, Cat.no. D000521) and Center for Disease Control (CDC; Hubei, China). 6-8 weeks old female NSG mice (for EV injection and MDA-MB-231 tumor models) or female BALB/c mice (for 4T1 tumor models) and female BALB/c-Nude mice were used in all studies and at least 5 mice per group. Mice were bred under specific pathogen free (SPF) condition, under 12 h light/12 h dark cycle (on at 6:00/off at 18:00 pm) with temperature of 72 ± 2 °F and 30–70% humidity. The experimental group and control mice were bred separately. For EV treatment, EVs were injected into the tail vein semiweekly for 5 weeks (~10 μg EVs per injection per mouse). For in vivo tracking of GFP- labeled EVs, EVs derived from MCF-10A/Lck-GFP, MDA-MB-231/Lck-GFP were injected into the tail vein for three consecutive days; mice were euthanized 6 h after the last injection and liver were collected for sectioning. Xenograft tumors were established by injecting 2 × 105 of MDA-MB-231 or 4T1 cells mixed with Matrigel (Corning) into the No.4 mammary fat pad. MDA-MB-468, MCF-7 and MDA-MB-231-LM2 were orthotopically injected into the No.4 mammary fat pad with 106 cells and Matrigel in nude mice, respectively. Before establishing the MCF-7 tumor, mice were subcutaneously injected with 40 µL of estradiol valerate (1 mg/mL) for two days. Mice received estradiol at 2, 5, 9, 13, 17, 21 and 25 days after cell injection. Tumor growth was monitored starting from 1 to 2 weeks after tumor cell injection, and tumor volume was calculated every other day using the formula (length × width2)/2. The maximum diameter of the tumor does not exceed 1.5 cm according to the IACUC. For restoration assay in liver, 5 × 1011 genomic copy of AAV8-mInsig1, AAV8-mInsig2 and AAV8-mATF3 virus or control virus were injected through the tail vein 1 week before implantation of MDA-MB-231 cells.
For 25-HC treatment, 231/Ctrl or 231/miR-9 KO mice were established by injecting 2 × 105 of wild type or miR-9-5p KO cells mixed with Matrigel into the number 4 mammary fat pad. When the volumes of xenograft tumors reached an average of 100 mm3, mice were randomly divided into control group and 25-HC group. For 25-HC group, mice were intraperitoneally injected with 25-HC (10 mg/kg) every other day until euthanized. For control group, mice were received the same volume of NaCl. For CH25H knockdown in liver, 5 × 1011 genomic copy of AAV-shCH25H virus or control virus were injected through the tail vein. One week after AAV injection, 4T1 cells were injected into the mammary fat pad to establish xenograft tumors. For desmosterol treatment, 4T1 cells were established into the number 4 mammary fat pad. Desmosterol (3 μg per mouse) was injected intraperitoneally every 2 days after tumor formation. For macrophage depletion in liver, MDA-MB-231 or 4T1 cells tumor bearing mouse were injected intravenously with clodronate liposomes 150 μL (SunLipo NanoTech) per mouse firstly, followed by 100 μL every 10 days. The mice were then euthanized and liver was collected for imaging and analysis of Kupffer cells. Oxygen consumption (VO2), carbon dioxide production (VCO2), heat and respiratory exchange ratio (RER) were measured concurrently using a 12 station Comprehensive Lab Animal Monitoring System (Columbus Instrument). Mice were placed in the metabolic cages 24 h before starting the experiment. At the start of the recording, measurements were taken at 12 min intervals. Ch25h-floxed mice were generated by Shanghai Model Organisms Center, Inc. using the CRISPR–cas9 system. Cas9 mRNA, single guide RNAs (sgRNAs) and donor were co-injected into zygotes. sgRNAs directed cas9 endonuclease cleavage in both sides of the Ch25h gene target exon 1, resulting in LoxP sites inserted, respectively, by homologous recombination. To obtain liver-conditional knockout mice (Ch25hL–/–), the floxed Ch25h mice (C57BL/6Smoc-Ch25hem1(flox)Smoc; Shanghai Model Organisms Center, Inc. Cat.no. NM-CKO-215043) were crossed with albumin-cre transgenic mice (B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J; Jackson Laboratory. Cat.no. 003574). Xenograft tumors were established by injecting 1 × 106 of E0771 cells mixed with Matrigel into the No.4 mammary fat pad. Before taking PET/CT, tumor cells (5 × 105 cells per injection) were re-suspended in 100 μL PBS and infused into the recipient mice by tail vein injection, followed by imaging. For miR-9-5pantagomir treatment, MDA-MB-231/Luc cells or 4T1/Luc cells were established into the number 4 mammary fat pad in NSG or BALB/c mice respectively. After tumor formation, NSG or BALB/c mice were randomly divided into two groups. MiR-9-5p antagomir or control (RiboBio) was directly injected into the implanted tumor at a dose of 10 nmol (dissolved in 50 μL PBS) every 3 days in NSG mice at the later stage of tumor growth or BALB/c mice at the later stage of tumor removal by tail vein injection. The mice were euthanized after 38 days and were collected for subsequent analysis. For lung, liver and colon cancer models, 1 × 106 of A549 cells, Hep3B cells and SW480 cells suspended in 100 μL PBS were injected subcutaneously into the right flank of nude mice. In all experiments, mice were euthanized by cervical dislocation.
Micro-PET/CT
Before 18F-FDG administration, mice were deprived of food for 8–12 h. 18F-FDG (5.5 MBq (150 μCi) in 0.1 mL) was injected through the tail vein. Micro-PET/CT imaging was started 1 h after 18F-FDG injection. Micro-PET/CT images was performed on a PET/CT scanner InliView-3000B (Novel Medical, Beijing, China). For micro-PET/CT imaging, all mice were subjected to isoflurane anesthesia (1–2% in 100% oxygen) on a temperature self-adjust tube to maintain body temperature throughout the procedure. Mice were visually monitored for breathing and any other signs of distress throughout the full imaging period. The images were reconstructed using a three-dimensional ordered subsets expectation maximum algorithm (OSEM) without attenuation correction. For data analysis, the region of interest (ROI) was manually drawn to cover the whole liver and lung on fused images.
Bioluminescence imaging
Bioluminescence imaging (BLI) of the luciferase activity in tumor cells was used to monitor the tumor metastasis progression. Before performing the BLI, the mice were anesthetized under sodium pentobarbital (100 mg/kg) and administered with D-luciferin (150 mg/kg) by intraperitoneal (i.p.) injection 10 min before the imaging. At the end time point, the liver and lungs were harvested in a 60 mm dish plate in 15 mg/ml D-luciferin diluted in PBS for bioluminescence imaging. Image exposure times were between 10 s and 1 min, depending on the signal strength. Light emission from the region of interest was quantified as photons s−1 cm−2 steradian−1.
Proteome profiler mouse chemokines array
The protein array was carried out using a proteome profiler mouse chemokines array kit (ARY020, R&D Systems, Minneapolis, USA). Two identical membranes precoated with antibodies against 25 different chemokines were incubated with media derived from 25-HC treated or control-RAW264.7 cells for 30 h, respectively. The following procedure was performed according to the manufacturer’s instructions. The semi-quantification of the dots was analyzed by ImageJ software.
Human serum specimens
Archived samples from patients with cancer patients and health controls used in this study were collected in accordance with and approved by Clinical Research Ethical Committee of Renmin Hospital of Wuhan University. All participants provided written informed consent. Archived fasting serum samples were collected from female patients with BC and female control subjects without cancer. The clinical samples from female patients with BC and control volunteers were obtained from Renmin Hospital of Wuhan University. Serum EVs were isolated using size exclusion chromatography (SEC)61. Briefly, samples were thawed at 37 °C and centrifuged at 3000 × g and room temperature for 10 min. The supernatant (500 µL) was applied onto a SEC column (qEV original 70 nm, Izon Science) and eluted by progressive addition of freshly filtered phosphate buffered saline (PBS), which allowed for separation and collection of 3 distinct 1.5 mL fractions where the intermediate tube contained EVs. In addition, serum EVs for small RNA sequencing are isolated using EXODUS62 (Shenzhen Huixin Life Technology). Trace plasma samples (20 µL) were diluted in 5 mL with 1x PBS and filtered through a 0.22 µm syringe filter (Millipore). The filtered plasma solutions were then loaded into the EXODUS device. Protein fragments, lipids, nucleic acids, and other small impurities pass through nanopores under the alternating negative pressing with EVs (diameter>20 nm) remaining inside the chamber. The EVs were then resuspended in 1x PBS up to 200 µL. All the clinical information was seen in Supplementary Table 7.
Software
Equipment build-in software were used for data collection, including the CFX Manager Software version 2.0 for real-time PCR data; Prism 9 was used for most data analysis and statistics; GSEA 4.3.2 was used for gene set enrichment analysis of RNA-seq data; http://cistrome.org/db/#/ is used to predict transcription factors for M2 marker genes. CytoFLEX2.4 was used for flow cytometry analysis for cells and characterization for EVs.
Statistics and reproducibility
All quantitative data are presented as mean ± standard deviation unless otherwise stated. All statistical analyses were performed using GraphPad Prism 9 (GraphPad Software). Two-tailed Student’s t tests were used for comparison of means of data between two groups. For multiple independent groups, one-way or two-way ANOVA with post hoc Tukey tests were used. Values of P < 0.05 were considered significant. Sample size was generally chosen based on preliminary data indicating the variance within each group and the differences between groups. Pearson correlation analysis was employed for analyzing the strength and direction of the linear relationship between two variables and represented as R value. All samples that have received the proper procedures with confidence were included for analyses. Animals were randomized before treatments. Western blots are repeated independently three times with similar results, and representative images are shown. The tissue slice contained three independent samples and representative images are shown.
Data availability
The RNA-seq data generated in this study has been deposited in the NCBI Gene Expression Omnibus (GEO) under accession code GSE222708 which includes GSE222622 (mice liver tissue receiving EV injection model), GSE222707 (mice liver tissue from xenografted tumor model) and GSE222623 (25-HC and vehicle treated RAW264.7 cells). The flow cytometry data has been deposited in Figshare (https://figshare.com/articles/online_resource/FACS/27537747). LC-MS/MS-detected oxysterols are summarized in Supplementary Data 1. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 71, 7–33 (2021).
Minn, A. J. et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005).
Kim, M. Y. Breast cancer metastasis. Adv. Exp. Med. Biol. 1187, 183–204 (2021).
Li, P. et al. Lung mesenchymal cells elicit lipid storage in neutrophils that fuel breast cancer lung metastasis. Nat. Immunol. 21, 1444–1455 (2020).
Nguyen, D. X., Bos, P. D. & Massague, J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284 (2009).
Valastyan, S. & Weinberg, R. A. Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013).
Coffelt, S. B. et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 (2015).
Nasrollahzadeh, E., Razi, S., Keshavarz-Fathi, M., Mazzone, M. & Rezaei, N. Pro-tumorigenic functions of macrophages at the primary, invasive and metastatic tumor site. Cancer Immunol. Immunother. 69, 1673–1697 (2020).
Dupuy, F. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015).
Lu, X. & Kang, Y. Organotropism of breast cancer metastasis. J. Mammary Gland Biol. Neoplasia 12, 153–162 (2007).
Zhou, W. et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer cell 25, 501–515 (2014).
Tkach, M. & Thery, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 164, 1226–1232 (2016).
Becker, A. et al. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 30, 836–848 (2016).
Redzic, J. S., Balaj, L., van der Vos, K. E. & Breakefield, X. O. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol. 28, 14–23 (2014).
Cao, M. et al. Cancer-cell-secreted extracellular vesicles suppress insulin secretion through miR-122 to impair systemic glucose homeostasis and contribute to tumour growth. Nat. Cell Biol. 24, 954–967 (2022).
Yan, W. et al. Cancer-cell-secreted miR-122 suppresses O-GlcNAcylation to promote skeletal muscle proteolysis. Nat. Cell Biol. 24, 793–804 (2022).
Marar, C., Starich, B. & Wirtz, D. Extracellular vesicles in immunomodulation and tumor progression. Nat. Immunol. 22, 560–570 (2021).
Lener, T. et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J. Extracell. Vesicles 4, 30087 (2015).
Fochtman, D. Taking risks. J. Pediatr. Oncol. Nurs. 8, 149–150 (1991).
Fong, M. Y. et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 17, 183–194 (2015).
Snaebjornsson, M. T., Janaki-Raman, S. & Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 31, 62–76 (2020).
Currie, E., Schulze, A., Zechner, R., Walther, T. C. & Farese, R. V. Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 (2013).
Simons, K. & Ikonen, E. How cells handle cholesterol. Science 290, 1721–1726 (2000).
Luo, J., Yang, H. & Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 21, 225–245 (2020).
Huang, B., Song, B. L. & Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2, 132–141 (2020).
Nelson, E. R., Chang, C. Y. & McDonnell, D. P. Cholesterol and breast cancer pathophysiology. Trends Endocrinol. Metab. 25, 649–655 (2014).
Chen, Y. Y., Ge, J. Y., Zhu, S. Y., Shao, Z. M. & Yu, K. D. Copy number amplification of ENSA promotes the progression of triple-negative breast cancer via cholesterol biosynthesis. Nat. Commun. 13, 791 (2022).
Ostrowski, M. et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 12, 19–30 (2010).
Gold, E. S. et al. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J. Exp. Med 209, 807–817 (2012).
Lund, E. G., Kerr, T. A., Sakai, J., Li, W. P. & Russell, D. W. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J. Biol. Chem. 273, 34316–34327 (1998).
Honda, A. et al. Cholesterol 25-hydroxylation activity of CYP3A. J. Lipid Res. 52, 1509–1516 (2011).
Patras, L., Shaashua, L., Matei, I. & Lyden, D. Immune determinants of the pre-metastatic niche. Cancer Cell 41, 546–572 (2023).
Lan, J. et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 79, 146–158 (2019).
Lu, Z. et al. ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. 34, 1342–1358.e1347 (2022).
Xu, J. et al. MiR-9 reduces human acyl-coenzyme A:cholesterol acyltransferase-1 to decrease THP-1 macrophage-derived foam cell formation. Acta Biochim. Biophys. Sin. (Shanghai) 45, 953–962 (2013).
Bottemanne, P. et al. 25-Hydroxycholesterol metabolism is altered by lung inflammation, and its local administration modulates lung inflammation in mice. FASEB J. 35, e21514 (2021).
Wculek, S. K. & Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015).
Tyagi, A. et al. Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nat. Commun. 12, 474 (2021).
Gong, Z. et al. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity 55, 1483–1500.e1489 (2022).
Gong, Z. et al. Lipid-laden lung mesenchymal cells foster breast cancer metastasis via metabolic reprogramming of tumor cells and natural killer cells. Cell Metab. 34, 1960–1976.e1969 (2022).
Dawson, S. J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209 (2013).
Bekes, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Li, M. X. et al. Degradation versus inhibition: development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Med Chem. 63, 4908–4928 (2020).
Charni-Natan, M. & Goldstein, I. Protocol for primary mouse hepatocyte isolation. STAR Protoc. 1, 100086 (2020).
Nnalue, N. A., Shnyra, A., Hultenby, K. & Lindberg AA. Salmonella choleraesuis and Salmonella typhimurium associated with liver cells after intravenous inoculation of rats are localized mainly in Kupffer cells and multiply intracellularly. Infect Immun. 60, 2758 (1992).
Gray, J. T. & Zolotukhin, S. Design and construction of functional AAV vectors. Methods Mol Biol. 807, 25 (2011).
Yan, W. et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 20, 597–609 (2018).
Fong, M. Y. et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 17, 183–194 (2015).
Kowal, J. et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 113, E968–E977 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12, 323–323 (2011).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Honda, A., Yamashita, K., Miyazaki, H., Shirai, M. & Matsuzaki, Y. Highly sensitive analysis of sterol profiles in human serum by LC-ESI-MS/MS. J. Lipid Res. 49, 2063–2073 (2008).
Fereidouni, F., Bader, A. N. & Gerritsen, H. C. Spectral phasor analysis allows rapid and reliable unmixing of fluorescence microscopy spectral images. Opt. Express 20, 12729–12741 (2012).
Cheng, J. X., Volkmer, A., Book, L. D. & Xie, X. S. Multiplex coherent anti-stokes Raman scattering microspectroscopy and study of lipid vesicles. J. Phys. Chem. B 106, 8493–8498 (2002).
Yue, S. H., Cardenas-Mora, J. M., Chaboub, L. S., Lelievre, S. A. & Cheng, J. X. Label-free analysis of breast tissue polarity by Raman imaging of lipid phase. Biophys. J. 102, 1215–1223 (2012).
Stranska, R. et al. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med 16, 1 (2018).
Chen, Y. et al. Exosome detection via the ultrafast-isolation system: EXODUS. Nat. Methods 18, 212–218 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32270827) and the Ministry of Science and Technology of China (2021YFA0804803) for W.Y. and (2018YFE0110800) for L.M.Z. We thank Dr. Pei-Shan Li from Soochow University for supplying E0771/Luc cells and Bao-Liang Song from Wuhan University for sharing immortalized bone-marrow-derived macrophages (iBMDMs) cells and their helpful advice. We thank Dr. Jun Chen from Sun Yat-sen University for his useful suggestions. We thank all the core facilities including microscopy core and electron microscopy facility from College of Life Science, ultracentrifugation platform provided by the State Key Laboratory of Virology and the pathology core from Renmin hospital of Wuhan University.
Author information
Authors and Affiliations
Contributions
W.Y. is the primary contact for this paper. W.Y. conceived ideas, W.Y. and L.M.Z. contributed to project planning. W.Y., M.X.L. and S.H. designed and performed most of the experiments. H.H.L. and L.M.Z. assisted with mass spectrometry analysis. J.J.L. provided clinical samples. H.J.L. and X.J.H. assisted with SRS imaging of liver tissue. X.H.Z. assisted with electron microscopy. M.Y., W.K.H., X.L., W.Q.J. and X.T.W. assisted with cell treatment and mouse experiments. B.W.X. and T.X.Y. assisted with cell line construction. D.F. assisted with liver- specific knockout CH25H mice. W.Y., M.X.L. and S.H. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, MX., Hu, S., Lei, HH. et al. Tumor-derived miR-9-5p-loaded EVs regulate cholesterol homeostasis to promote breast cancer liver metastasis in mice. Nat Commun 15, 10539 (2024). https://doi.org/10.1038/s41467-024-54706-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54706-z
