
Abstract
Developing efficient and stable oxygen evolution reaction electrocatalysts under acidic conditions is crucial for advancing proton-exchange membrane water electrolysers commercialization. Here, we develop a representative strategy through p-orbital atoms (N, P, S, Se) doping in RuO2 to precisely regulate the lattice oxygen-mediated mechanism-oxygen vacancy site mechanism pathway. In situ and ex situ measurements along with theoretical calculations demonstrate that Se doping dynamically adjusts the band gap between the Ru-eg and O-p orbitals during the oxygen evolution reaction process. This modulation accelerates electron diffusion to the external circuit, promotes the lattice oxygen-mediated process, and enhances catalytic activity. Additionally, it facilitates electron feedback and stabilizes oxygen vacancies, thereby promoting the oxygen vacancy site mechanism process and enhancing catalytic stability. The resulting Se-RuOx catalyst achieves efficient proton-exchange membrane water electrolysers performance under industrial conditions with a minimal charge overpotential of 1.67 V to achieve a current density of 1 A cm−2 and maintain long-term cyclability for over 1000 h. This work presents a unique method for guiding the future development of high-performance metal oxide catalysts.
Similar content being viewed by others

Introduction
The oxygen evolution reaction (OER) plays an indisputably critical role in energy conversion systems, underpinning processes such as hydrogen production via water electrolysis, CO2 reduction to generate clean small-molecule fuels, and the utilization of energy conversion technologies such as metal‒air batteries1,2,3. As an essential half-reaction, the OER requires more energy to surmount the kinetic barrier owing to its sluggish four-electron transfer process, which is a bottleneck for realizing the industrialization of those conversion technologies4,5,6. Noble metal Ir-based nanomaterials are currently regarded as the most effective catalysts for the acidic OER, but their scarcity and exorbitant cost pose significant challenges to widespread implementation7,8,9,10. Moreover, owing to their high activity and relatively low cost (approximately one-sixth of the price of Ir), Ru-based nanomaterials hold tremendous potential in the field of proton-exchange membrane water electrolysers (PEMWEs), but the stability of RuO2 in acidic environments remains unsatisfactory10,11,12. To increase the overall efficiency of PEMWEs, it is necessary to develop efficient and stable Ru-based catalysts for the oxygen evolution reaction in acidic media.
Ru-based catalysts, which tend to follow the lattice oxygen oxidation mechanism (LOM) pathway, can bypass the typical thermodynamic constraints of the traditional adsorption evolution mechanism (AEM) to increase OER kinetics13,14,15. However, the persistent involvement of lattice oxygen (OL) during the continuous LOM pathway (LOM-LOM) can lead to excessive oxidation of Ru and its dissolution into the electrolyte, resulting in poor stability16,17,18,19. Currently, transforming the LOM pathway of Ru-based catalysts to the AEM pathway to suppress the emergence of high-valence Run>4+ species is considered an effective means to improve the stability of Ru-based catalysts, but the OER activity is inevitably constrained by the thermodynamic linear relationship13,20,21. Thus, combining the advantages of the AEM and LOM, i.e., promoting catalytic activity by initiating the OER through lattice oxygen and improving stability by inhibiting the continuous participation of lattice oxygen in the OER, is an effective strategy for designing high-performance Ru-based catalysts. In an exciting recent breakthrough, the lattice oxygen-mediated mechanism-oxygen vacancy site mechanism (LOM-OVSM) was proposed, which could bypass the linear relationship of oxygen intermediates and avoid the generation of Run>4+, thereby enabling the activities and stabilities of the OER to be even superior to the theoretical limit, offering a promising advanced pathway for the design of improved Ru-based catalysts22,23,24. As demonstrated in the pioneering research reported by Wang et al., an electrocatalyst satisfying the requirements for the LOM-OVSM-type OER can potentially overcome the so-called scaling limit without sacrificing stability25. During the LOM-OVSM pathway, the LOM-mediated process can increase the kinetic rate, and the OVSM process can prevent the formation of excessive high-valence Ru species, which requires the rapid diffusion of electrons to the external circuit and their swift return. However, the lack of comprehensive research on the relationship between the electronic structure of Ru-based materials and the OER mechanism may inevitably lead to additional challenges in constructing the LOM-OVSM pathway for Ru-based catalysts. Owing to the spatial overlap between orbitals and similar energy distributions of electronic states, d–p orbital hybridization naturally occurs following the introduction of light elements (F, C, O, N, P, S, Se, etc.) and provides substantial flexibility for optimizing the electronic structure, thereby contributing to high catalytic performance26,27. Therefore, a more in-depth understanding of the electronic structure‒mechanism relationship can be established by providing an unequivocal structural analysis of Ru-based materials with appropriate doped anions; however, research on this topic is limited.
In this work, we pioneer the doping of different p-orbital atoms R (N, P, S, Se) into RuO2 (R-RuOx) with a simple one-step calcination synthesis method to investigate the electronic structure‒mechanism relationship of Ru-based materials and the construction principle of the LOM‒OVSM path. In situ and ex situ measurements along with theoretical calculations demonstrated that the notable OER activity and stability of Se-RuOx originate from the dynamic adjustment of the band gap between the Ru-eg and O-p orbitals during the OER process. Notably, Se-RuOx catalyst also translates into promising performance in PEMWE devices, exhibiting a minimal charge overpotential of 1.67 V to deliver 1 A cm−2 and maintaining cyclability for over 1000 h. Overall, a Se-RuOx catalyst that dynamically adjusts the band gap between the Ru-eg and O-p orbitals is essential for creating LOM-OVSM pathway catalysts that notably enhance both OER activity and stability, presenting a promising avenue for the strategic development of high-performance metal oxide catalysts.
Results
Materials characterization
X-ray diffraction (XRD) measurement was conducted to identify the phase information for all the catalysts. As shown in Fig. 1a, b, all the XRD patterns display the same group of peaks in agreement with the RuO2 phase (JCPDS No. 40-1290). Moreover, with the introduction of R (R = N, P, S, or Se), the peak positions of R-RuOx tend to shift towards lower angles than those of RuO2, without the appearance of any additional diffraction peaks, indicating the successful doping of N, P, S, or Se without damaging the structure. The transmission electron microscopy (TEM) images shown in Fig. 1c and Figs. S1–5 demonstrate the flexible nanoparticle morphology of the as-prepared R-RuOx with smooth and evenly dispersed surfaces. Compared with that of RuO2, the lattice spacing of R-RuOx slightly expands, further indicating the successful introduction of anionic R, which is consistent with the XRD results. Then, spherical aberration-corrected (HAADF-STEM) was employed to investigate the Se atom distribution in the Se-RuOx hybrid nanostructures. Energy-dispersive X-ray spectroscopy (EDS) elemental mapping images revealed a uniform distribution of all the elements within Se-RuOx (Fig. 1d). Se-RuOx nanoparticles with an average diameter of 10 nm were further determined from HAADF-STEM images, as shown in Fig. 1e and Fig. S6. The high-resolution TEM images obtained via spherical aberration correction show interplanar spacings of 0.32 and 0.26 nm, which correspond to the (110) and (101) lattice fringes of face-centred cubic (FCC) RuO2 (Fig. 1f–h). This suggests that Se-RuOx closely resembles the crystal structure of RuO2, with Se atoms exclusively located at the O position, and the corresponding crystal structure model of Se-RuOx simulates consistent element spread (Fig. 1i).

a XRD patterns of RuO2 and R-RuOx (R = N, P, S, Se). b Extended XRD patterns derived from (a). c TEM images of Se-RuOx. d Elemental mappings of Se-RuOx at a 1 nm scale by HAADF-STEM. e–g AC-HAADF-STEM images and the corresponding lattice fringe profile of Se-RuOx. h FFT pattern in (g). i HAADF-STEM image and the simulated molecular structure of Se-RuOx.
X-ray absorption spectroscopy (XAS) techniques were utilized to investigate the electronic configuration of R-RuOx catalysts, providing valuable insights into the lattice oxygen activity and OER catalytic pathways in R-RuOx catalysts. X-ray absorption near-edge structure spectroscopy (XANES) of the Ru K edge, as shown in Fig. 2a, b, reveals variations in the adsorption threshold position and oxidation state of the R-RuOx catalyst with different R values. Se-RuOx has the lowest valence state, indicating the highest covalency of the Ru-O bond and the most electron-rich state in Se-RuOx, thereby facilitating the LOM process28,29. Furthermore, Ru K-edge Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectroscopy was used to investigate the local coordination environment of R-RuOx. The main peak at 1.967 Å for pure RuO2 corresponds to the first Ru-O coordination shell of the Ru cations (Fig. 2c)30,31. Compared with those of RuO2, increases in the length of the Ru-O bond are observed in Se-RuOx (1.978 Å), S-RuOx (1.976 Å), P-RuOx (1.971 Å) and N-RuOx (1.973 Å), suggesting the enhanced reactivity of lattice oxygen, particularly in Se-RuOx (Figs. 2c, d, S7–8 and Table S1). Furthermore, wavelet transform analysis corroborated the results from the FT-EXAFS spectra and revealed the longest Ru-O bond length in Se-RuOx (Fig. 2e). Additionally, the O K-edge XANES spectrum for R-RuOx catalysts, as shown in Fig. 2f, reveals two distinct sharp peaks at 531.7 and 534.7 eV, corresponding to the excitation of O 1 s core electrons into hybridized states between O 2p and Ru 4 d t2g and eg states caused by the splitting of an octahedral field32,33,34. Another broad peak at higher energy is attributed to the hybridization of the O 2p orbital with Ru 5sp states35. Compared with that of RuO2, the increased t2g/eg intensity further reveals the increased Ru-O covalency in R-RuOx, with Se-RuOx being particularly notable, indicating the highest degree of lattice oxygen activation in Se-RuOx and thus making it more conducive to promoting the formation of VO (Fig. 2g).

a Ru K-edge XANES spectra, (Inset: Partially enlarged image). b Oxidation state of ruthenium species obtained from Ru K-edge XANES. c Ru K-edge FT-EXAFS spectra. d The fitted Ru‒O bond length (horizontal axis R: coordination bond length). e Wavelet transform for Ru K-edge EXAFS signals (k: Wave vector number). f O K-edge XANES spectra. g t2g/eg intensities of RuO2 and R-RuOx. h Ru 3p XPS spectra. i O 1 s XPS spectra (Vo: oxygen vacancy; OL: Lattice oxygen; H2O: absorbs oxygen). j EPR spectra. k Inverse susceptibility 1/χ against temperature. l Effects of Se doping on the Ru-d orbital structure and the changes in the active centre (LOM lattice oxygen oxidation mechanism, LOM-OVSM lattice oxygen-mediated mechanism-oxygen vacancy site mechanism).
To verify the impact of R doping on the formation and stability of VO during the OER process, the electronic structures of R-RuOx after cyclic voltammetry (CV) activation (VO-R-RuOx) in the potential range of 1.1 to 1.6 V vs. RHE were investigated. First, X-ray photoelectron spectroscopy (XPS) was used to determine the VO concentration of VO-R-RuOx. The Ni 1 s, P 2p, S 2p and Se 3 d XPS spectra shown in Fig. S9 further demonstrate the successful introduction of anionic R (R = N, P, S, Se). Figure 2h shows that the Ru 3p spectra of all the VO-R-RuOx samples are almost identical, but the two peaks of Ru 3p3/2 and Ru 3p1/2 in VO-Se-RuOx shift towards lower binding energies than those of the other VO-R-RuOx samples do, indicating that VO-Se-RuOx possesses more low-valence Ru ions and VO. In the O 1 s spectra, the relative contribution of the peak related to VO in VO-Se-RuOx is 55.03%, which is markedly greater than that in the other samples, demonstrating a richer presence of VO in VO-Se-RuOx (Fig. 2i)36,37. Moreover, VO-Se-RuOx shows a stronger electron paramagnetic resonance (EPR) signal at g = 2.004, which also proves that VO-Se-RuOx contains more VO than other VO-R-RuOx (Fig. 2j)38. The results from XAS, XPS, and EPR analyses indicate that Se-RuOx is more prone to facilitate the LOM-mediated process and produce abundant VO. Additionally, the temperature-dependent magnetizations (M-T) curves for VO-R-RuOx were collected with a magnetic field of H = 500 Oe under field-cooling procedures to probe the spin structures of the Ru centres because of the high sensitivity of magnetism to unpaired electrons (Fig. S10). The magnetic susceptibilities above 150 K obey a paramagnetic Curie–Weiss law, and the eg-electron filling state of the sample can be calculated by fitting39. As expected, an impressive relationship with the trend of eg-electron filling state after activation, VO-Se-RuOx (1.16) > VO-S-RuOx (0.95) > VO-P-RuOx (0.73) > VO-N-RuOx (0.54) > VO-RuO2 (0.43), can be observed (Fig. 2k, and Table S2), demonstrating faster electron compensation from the outer circuit back to the eg orbitals of VO-Se-RuOx, thus eliminating the overoxidation of Ru and promoting the OVSM process40. In summary, as shown in Fig. 2l, when O atoms are replaced by Se atoms, the dz2 (eg) energy decreases, allowing electrons from the O-p orbital to transfer more rapidly to the eg orbital and subsequently to the external circuit, thereby enhancing the LOM-mediated process. After the LOM pathway occurs, the electrons of VO-Se-RuOx return quickly, thereby stabilizing the VO, leading to increased energy of the t2g orbital and decreased energy of the O-p orbital, thereby promoting the OVSM process41,42.
Electrochemical and spectroscopic techniques were further utilized to identify the reaction kinetics, validating the potential LOM-OVSM pathway as considered above, which facilitates deciphering the notable OER performance. Linear sweep voltammetry (LSV) curves of R-RuOx and RuO2 in H2SO4 (pH 0-1) at a scan rate of 5 mV s⁻¹ were recorded (Fig. 3a and S11). All the R-RuOx and RuO2 samples demonstrated obvious pH dependence, suggesting nonconcerted proton‒electron transfer (NCPET) processes7,43. Furthermore, good linear relationships were observed between the corresponding log(j) values at different potentials and the electrolyte pH values, emphasizing the mediated LOM pathway during the OER in R-RuOx and RuO2 (Fig. 3b). Notably, the pH correlation of Se-RuOx is much weaker than that of RuO2, indicating that Se-RuOx may not exclusively follow the LOM pathway during the OER process. In addition, to gain insight into the OER mechanism, the oxidation behaviour of Ru sites at high overpotentials was investigated via cyclic voltammetry (CV) analysis between 0.3 and 1.2 V vs. SCE at a scan rate of 10 mV/s for R-RuOx and RuO2 (Fig. 3c). Two groups of redox peaks at approximately 0.72 and 0.98 V could be observed for RuO2, which could be assigned to Ru3+/Ru4+and Ru4+/Ru6+, respectively44,45. The reduction peak at 1.12 V corresponds to Ru6+/Ru8+ and is attributed to the excessive accumulation of the metastable dissolution intermediate *RuO42- (continuous LOM pathway)46. Compared with those of other R-RuOx materials, the peaks of Ru3+/Ru4+ in Se-RuOx are more dominant than those in Ru4+/Ru6+, and the peak potentials of Ru3+/Ru4+ (0.61 V) and Ru4+/Ru6+ (0.91 V) are lower, indicating that Se-RuOx has rapid electron diffusion capability, thereby improving the OER activity9. Furthermore, the peak of Ru4+/Ru6+ in Se-RuOx exhibited a distinctly decreased area accompanied by the disappearance of the redox peak for Ru6+/Ru8+, indicating that Se doping promoted electron feedback and suppressed the overoxidation of Ru sites, thereby improving OER stability. Furthermore, in situ Raman spectroscopy was employed to probe the surface state of Se-RuOx and RuO2 during the OER. As depicted in Fig. 3d, three main Raman features of rutile RuO2 can be observed on both Se-RuOx and RuO2, including the Eg, A1g and B2g vibration modes47. In addition, when the applied potential exceeds 1.20 V (Se-RuOx) or 1.40 V (RuO2), a Raman band can be found at approximately 640 cm−1, which can be assigned to the presence of Vo, demonstrating that Se-RuOx can form Vo more rapidly during the OER48,49. Importantly, when the applied voltage exceeds 1.35 V, another Raman band belonging to *OOH (791 cm−1) appears in Se-RuOx, further indicating that Se-RuOx does not follow the traditional LOM pathway during the OER but rather follows the LOM-OVSM pathway. To further confirm that the LOM-OVSM pathway is involved in Se-RuOx during the OER process, operando 18O isotope labelling differential electrochemical mass spectrometry (DEMS) measurements were performed (Fig. S12). The RuO2 and Se-RuOx were labelled by conducting CV (1.2-1.65 V vs. RHE) in H218O aqueous sulfuric acid electrolyte, followed by OER testing in the nonlabelled aqueous (H216O) sulfuric acid electrolyte. The results for both RuO2 (Fig. S13) and Se-RuOx (Fig. 3e) show that the main mass signal is 32O2, followed by 34O2, with no 36O2 signal. Compared with that of RuO2, the significant decrease in the ratio of 34O2 to 32O2 demonstrates that the majority of the intrinsic lattice oxygen in Se-RuOx does not participate in O‒O coupling, thereby excluding the traditional LOM24,25. In situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was employed to further identify the reaction intermediates during the OER. For RuO2 and N-RuOx, P-RuOx, and S-RuOx, an obvious absorption band emerges at ~1200 cm−1 at high overpotentials (Fig. 3f and Fig. S14), which is attributed to the stretching vibration of the *OO intermediate50. This indicates that the OER process of RuO2 and N-RuOx, P-RuOx, and S-RuOx is predominantly governed by continuous LOM at high overpotentials, consequently accelerating dissolution and diminishing stability. In contrast, the spectrum of Se-RuOx shows that the dynamic *OO absorption band appears at a lower voltage ( ~ 1.05 V), indicating the mediation of the LOM mechanism (Fig. 3g). Moreover, the vibrational band of the crucial *OOH intermediate (at ~1050 cm−1) in the Se-RuOx electrocatalyst emerges at ~1.15 V and notably intensifies with increasing potential, excluding the traditional LOM51. The combined findings from in situ ATR-SEIRAS, in situ Raman spectroscopy and DEMS indicate that Se-RuOx involves a unique LOM-OVSM pathway different from the LOM pathway of RuO2, which not only promotes the reaction kinetics but also inhibits the dissolution of the Ru-based catalyst.

a pH dependence of Se-RuOx. b The linear relationship of onset potential vs. pH. c CV analysis of the redox peaks of RuO2 and R-RuOx measured from 0.0 to 1.2 V vs. SCE. d In situ Raman spectra of RuO2 and Se-RuOx during the OER. e DEMS tests of 16O16O, 18O16O and 18O18O signals from the catalysts for 18O-labelled R-RuOx in 0.5 M H2SO4 in H216O. In situ ATR-SEIRAS spectra of f RuO2 and g Se-RuOx during the OER.
Electrocatalytic OER performance
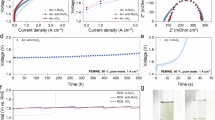
Encouraged by the unique LOM-OVSM pathway, we further surveyed potential applications for the acidic OER. The electrochemical OER performance of the R-RuOx electrocatalyst was assessed by a standard three-electrode system in a 0.5 M H2SO4 electrolyte. As shown in Fig. 4a, b and Fig. S15, Se-RuOx displays notable catalytic activity compared with the other R-RuOx electrocatalysts, reflecting the smallest required overpotential of 188 mV to deliver 10 mA cm−2, which is better than to the benchmarks of RuO2 (279 mV), N-RuOx (215 mV), P-RuOx (209 mV), S-RuOx (201 mV) and most of the Ru-based catalysts reported to date (Table S3). In addition, Se-RuOx has a lower Tafel slope (64.6 mV dec−1) than RuO2 (80.2 mV dec−1), S-RuOx (65.2 mV dec−1), P-RuOx (66.4 mV dec−1) and N-RuOx (76.5 mV dec−1) (Fig. 4c). The lower Tafel slope reveals more facile OER kinetics for Se-RuOx, and the potential-determining step (PDS) tends towards the *O-OH coupling step during the OVSM pathway, indicating that the Se-doping system can accelerate the corresponding electron reaction. Notably, Se-RuOx exhibited the highest turnover frequency (TOF) value among R-RuOx at 1.55 V vs. RHE, indicating its enhanced intrinsic electrocatalytic activity (Fig. S16). Moreover, Se-RuOx displays a lower charge transfer resistance (Rct), which is less than to that of other R-RuOx, as evidenced by a smaller Nyquist cycle diameter from electrochemical impedance spectroscopy (EIS) measurements (Fig. S17 and Table S4). Furthermore, Additionally, in situ EIS analyses at various applied potentials were utilized to survey the electrocatalytic kinetics occurring during OER (Figs. S18-19). In the Bode phase diagrams, the observed maxima at elevated frequencies demonstrate the electrocatalyst’s inherent electron transport properties, while corresponding peaks detected in lower frequency domains characterize interfacial charge transfer processes between electrolyte and catalyst materials52,53. The observed electrochemical behavior in Fig. 4d, e and Fig. S20 demonstrates that R-RuOx catalysts consistently display amplified phase signal intensity within the low-frequency ___domain ( ≈ 0.01-10 Hz) relative to their high-frequency regions ( ≈ 100—10000 Hz). This frequency-dependent response suggests that interfacial charge transfer mechanisms are principally governed by resistive interactions at the electrolyte‒catalyst interface. As operational voltage escalates from 1.25 V to 1.55 V, pronounced phase angle attenuation emerges specifically in the low-frequency regime of Se-RuOx, contrasting with other R-RuOx materials, which demonstrates that Se doping can reduces interfacial charge transfer resistance between electrocatalyst and electrolyte, resulting in optimized electron transport efficiency that directly improves oxygen evolution reaction performance. In addition, the electrochemical surface area (ECSA) was determined through the calculation of the double-layer capacitance (Cdl). As shown in Fig. S21, the Cdl of Se-RuOx is 64.7 mF cm−2, which is close to those of RuO2 (36.3 mF cm−2),S-RuOx (57.8 mF cm−2), P-RuOx (46.7 mF cm−2) and N-RuOx (44.2 mF cm−2), indicating the greater number of exposed active sites in Se-RuOx. To elucidate the TOF progression pattern undetected in conventional polarization measurements (Fig. S16), the voltage polarization curves normalized by ECSA were constructed. (Fig. S22). Interestingly, a comparable trend to that identified in the TOF calculations is evident, clearly indicating the combined role of the intrinsic catalytic characteristics on determining the practical performance of Se-RuOx. Given the limited stability of traditional RuO2 under acidic oxygen evolution environments, the stability assessment of the synthesized Se-RuOx was investigated in 0.5 M H2SO4. Se-RuOx exhibits fully retained activity following 1000 CV cycles relative to its original state (Fig. 4f). In comparison, a significant increase in the overpotential is found for the com. RuO2 electrocatalyst (Fig. S23). Furthermore, Se-RuOx exhibited nearly unaltered OER performance during the chronopotentiometric test over a period of approximately 150 h at 100 mA cm−2, which was much better than that of the other R-RuOx ( < 10 h) (Fig. 4g, h). ICP-MS test was further performed to quantify the dissolution rates of Se and Ru. As illustrated in Figs. S24-25 and Table S5, Se-RuOx demonstrates notably low dissolution rates of 0.91% for Ru and 2.29% for Se after 100 h stability testing, with the stability number (S-number) determined to be 2.2*105, indicating its comparable long-term durability. In situ PXRD was performed to provide in-depth information and realistically assess the durability of Se-RuOx at different applied potentials (Fig. 4i). Se-RuOx retains its high crystallinity under OER operating potentials, further revealing its electrochemical stability.

a LSV curves of RuO2 and R-RuOx in a 0.5 M H2SO4 solution. b The overpotentials at 10 mA cm−2 and 100 mA cm−2 of RuO2 and R-RuOx. c Corresponding Tafel plots according to the LSV curves in (a). d Bode phase plots of Se-RuOx. e Summarized phase peak angles of RuO2 and R-RuOx at 1.25–1.55 V. f LSV curves of initial and after 1000 CV cycles on Se-RuOx. g Chronopotentiometric curves of RuO2 and R-RuOx at a current density of 10 mA cm−2 for 10 h. h Chronopotentiometric curves of Se-RuOx at a current density of 100 mA cm−2 for 150 h. i In situ XRD patterns of Se-RuOx at 1.3 V-1.8 V.
Theoretical insights into intrinsic activity and stability
Our experimental results verify that Se-RuOx changes the OER pathway from the traditional LOM to LOM-OVSM, thereby notably enhancing the OER performance. To determine the reasons behind the enhanced activity and stability of Se-RuOx and to understand why the LOM-OVSM pathway is favoured in Se-RuOx, density functional theory (DFT) calculations were carried out. Based on the classical theory, the metal site tends to bond with oxygen through hybridization between the p and d orbitals, resulting in d orbitals of active centres splitting into eg and t2g orbitals (Fig. 5a)54. When Se is substituted for O, the O-p orbitals upshift to Ef, and the dz2 (eg) orbitals move down, making it easier for electrons to transfer from the nonbonding O-p band to the eg orbitals and then diffuse to the external circuit, thereby promoting the kinetics of the LOM-mediated process. When abundant VO form during the OER process in the LOM pathway, Se doping can increase the proportion of empty eg orbitals, promote electron return, prevent the formation of high-valence Ru species and stabilize the VO. When stable vacancies form, the O-p orbital energy decreases, and the t2g orbital energy increases compared with that of RuO2, allowing electrons to more easily transfer from t2g to eg orbitals, thereby promoting the OVSM pathway (Fig. 5b). In particular, the introduction of an appropriate element is reasonable for broadening the eg band and upshifting the t2g orbitals, resulting in the conversion of the active centre from the O centre to the M centre (Fig. 5c) and conducting a unique LOM-OVSM route different from the LOM path of RuO2 (Fig. 5d)55.

a Ru-d orbital splitting and hybridization schematic. b Band structures of the compounds synthesized from RuO2 and Se-RuOx during the OER process. The position of the O2/H2O redox couple is 1.23 V versus RHE, as shown schematically on the right. The relationship between the voltage under the RHE and standard hydrogen electrode (SHE) scale is ƐRHE=ƐSHE + 59 mV×pH (pH=0 in 0.5 M H2SO4). c Schematic band structure of different active centres. d Adsorption/desorption and electron transfer processes of OER intermediates on the surface of the traditional LOM pathway (on RuO2 surface) and LOM-OVSM pathway (on Se-RuOx surface) (LOM lattice oxygen oxidation mechanism, LOM-OVSM lattice oxygen-mediated mechanism-oxygen vacancy site mechanism).
For the computational studies, we began by building R-RuOx (R = N, O, S, P, Se) models based on an established RuO2 compactional framework (Figs. S26, 27). First, the partial orbital density of states (PDOS) of Ru atoms and O atoms for R-RuOx samples were analysed and are presented in Fig. 6a and S28. The results show that the energy gap (∆ɛ) between the O-p band and the Ru-eg band follows the order RuO2 > N-RuOx > P-RuOx > S-RuOx > Se-RuOx, indicating increased lattice oxygen activity in Se-RuOx compared with other R-RuOx and RuO2 (Fig. 6b)56. This is further confirmed by the ∆ɛ between the O-p band and Ru-d band, which shows a similar trend (Fig. S29). Furthermore, the free energy profiles indicate that the OER on all R-RuOx materials follows the LOM pathway rather than the AEM pathway and that Se-RuOx has the highest OER kinetics (Fig. S30). Next, the structural properties following the formation of lattice oxygen (Vo-R-RuOx) during the LOM process were examined, thereby identifying the reasons for the disruption of the traditional LOM pathway (Figs. S31, 32). The PDOSs for the eg and t2g orbitals of the Ru site for the Vo-R-RuOx samples were analysed to verify the orbital structures in Fig. 2l and are presented in Fig. 6c and S33–37. In particular, Vo-Se-RuOx has the highest Ru-eg empty rate and the highest Ru-t2g band centre, as well as the smallest gap between the eg and t2g orbitals (Figs. 6c, d and S34, 35). This facilitates the electron transition from the t2g orbital to the eg orbital and promotes electron feedback from the external circuit to the eg orbital, thereby suppressing the continuous oxidation and dissolution of Ru. Moreover, the gap between the O-p band and the Ru-eg band increased from Vo-RuO2 to Vo-N-RuOx, Vo-P-RuOx, Vo-S-RuOx, and Vo-Se-RuOx (Fig. 6e, f and Fig. S36). The increased gap between the O-p band and the Ru-eg band in Vo-Se-RuOx promotes electron transfer from the t2g orbital to the eg orbital and then diffuses to the external circuit, converting the redox centre from the lattice oxygen to the metal centre, thereby transforming the traditional LOM pathway into the OVSM pathway. The lowest O-p band and the largest ∆ɛ between the O-p band and Ru-d band in Vo-Se-RuOx also confirm the transformation of redox centres from the lattice oxygen to the metal centre (Fig. S37). In addition, compared with other Vo-R-RuOx, Vo-Se-RuOx possesses the lowest Ru positive charge enrichment (Figs. 6g and S38), the regression of more Ru electrons (Fig. S39), the rapid electron returns to the surface of Ru atoms (electron localized function, ELF, Fig. S40) and the smallest band gap (Fig. S41), which all further confirm the above conclusions. As expected, the optimized LOM-OVSM path on Se-RuOx is favourable for changing and decreasing the rate-determining step (RDS) (*OH → *O) ( ~ 1.66 eV), which results in the lowest required RDS energy compared with that of RuO2 (1.73 eV), N-RuOx (1.71 eV), P-RuOx (1.73 eV) and S-RuOx (1.69 eV), indicating better reaction kinetics (Figs. 6h and S42)9. On the basis of these results, the notable OER activity and stability of Se-RuOx originate from the following. (1) The decreased ∆ɛ between the O-p band and Ru-eg bands promotes electron diffusion into the external circuit and then facilitates the initiation of the LOM pathway, thereby increasing the OER catalytic activity. (2) The increased ∆ɛ between the O-p band and Ru-eg band and eg orbital broadening promote electron return from the external circuit to avoid the formation of high-valence Ru species and change the traditional LOM path to OVSM, thereby improving OER stability.

a PDOS plots of the Ru-eg and O-p states in Se-RuOx. b Band centres of the Ru-eg orbital and O-p orbital in RuO2 and R-RuOx (R = N, S, P, Se). c PDOS plots of the Ru-eg and Ru-t2g states in VO-Se-RuOx. d Ru-eg empty rates of VO-RuO2 and VO-R-RuOx. e PDOS plots of the Ru-eg and O-p states in VO-Se-RuOx (Inset: the specific atoms on Vo-Se-RuO x surface). f Band centres of Ru-eg and O-p in VO-RuO2 and VO-R-RuOx. g Differential charge density of RuO2 and Se-RuOx. h Free energy profiles of different OER intermediates of VO-Se-RuOx.
In addition, DFT calculations for Se-RuOx with different doping sites indicate that Se doping along the z-direction can lead to the optimal ∆ɛ between the O-p band and Ru-eg bands and promote the LOM-OVSM pathway, thereby having the optimal OER performance (Figs. S43, 44). DFT calculations show that with a doping ratio of 1/12, the LOM-OVSM pathway occurs, resulting in increased OER activity and stability (Figs. S45–47). The electrocatalytic performance was assessed in 0.5 M H2SO4 utilizing a standard three-electrode system. As shown in Fig. S48, Se-RuOx-2 displays the best acidic OER activity, with an overpotential of 189 mV to achieve a current density of 10 mA cm−2, which is much lower than those of Se-RuOx-1 (238 mV), Se-RuOx-4 (219 mV) and Se-RuOx-6 (233 mV). Furthermore, with increasing Se doping ratio, the stability of the Ru-based catalyst initially increases but then decreases. An excessively high ratio of Se leads to accelerated dissolution of Ru, thereby reducing the stability of the catalyst, which is consistent with the DFT calculations.
PEMWE electrolyzer
Finally, we sought to operate the Se-RuOx catalyst in practical applications. A membrane electrode assembly (MEA)-based PEMWE was constructed with Se-RuOx as the anode catalyst, commercial Pt/C as the cathode catalyst, and Nafion®211 as the proton-exchange membrane (Fig. 7a and Fig. S49). As a benchmark, a similar PEMWE was fabricated using other R-RuOx as the anode catalyst. All device performances were evaluated in 0.5 M H2SO4 at 25 °C without iR correction. Compared with Fig. S50, to reach a water-splitting current density of 1 A cm−2, the Se-RuOx-based PEMWE required a full-cell voltage of 1.84 V, which was much lower than those of other R-RuOx-based PEMWE. In addition, the activity of Se-RuOx gradually increased with increasing temperature (Fig. 7b). When the temperature reaches 80 °C (the industrial level), the full-cell voltage of the Se-RuOx-based PEMWE is 1.59 V at 0.5 A cm−2, 1.67 at 1 A cm−2 and 1.74 V at 1.5 A cm−2, highlighting its competitive performance in PEM electrolysers. Moreover, the Faraday efficiencies for O2 generation were evaluated and showed excellent agreement with the theoretical values based on the O2 molar ratio, thereby confirming a Faraday efficiency approaching almost 100% (Fig. S51). More impressively, the Se-RuOx-based PEMWE demonstrated markedly improved device stability at 1 A cm−2 in 0.5 M H2SO4. As shown in Fig. 7c, the Se-RuOx-based PEMWE can operate stably for 1000 h, with a degradation rate of only 58 μV•h−1. The voltage of the RuO2-based PEMWE increased by 600 mV in just a few hours. In contrast, Se-RuOx demonstrates notable performance over the reported Ru-based OER catalysts in terms of the ampere-level current density and long-term stability (Table S6). These results underscore the notable OER activity and stability metrics of the Se-RuOx catalyst at an industrial-level current density in PEM devices.

a Schematic illustration of the designed PEMWE water electrolyzer. b LSV curves of the PEMWE water electrolyzer for Se-RuOx at 25, 40, and 80 °C. c Durability cell voltage‒time plots for the PEMWE water electrolyzer at a constant current density of 1000 mA cm−2 for 1000 h.
Discussion
In summary, we pioneer the doping of different p-orbital atoms (N, P, S, Se) into RuO2 (R-RuOx) with a simple one-step calcination synthesis method to investigate the electronic structure‒mechanism relationship of Ru-based materials and the construction principle of the LOM‒OVSM path. Extensive experimental and theoretical evidence has confirmed that when p-orbital Se atoms are doped, electrons are more likely to diffuse from the O-p orbitals to the Ru-eg orbital, accelerating their diffusion to the external circuit and resulting in a faster LOM-mediated process. When abundant oxygen vacancy (VO) forms during the LOM process, Se doping can induce an increase in Ru-eg band broadening, promoting electron return and stabilizing the VO, leading to increased energy of the Ru-t2g orbital and decreased energy of the O-p orbital, causing electrons to transfer from the Ru-t2g orbital to the Ru-eg orbital and thereby promoting the OVSM process. As a result, the representative Se-RuOx exhibited a low overpotential (188 mV at 10 mA cm−2) and stable PEMWE performance (1.67 V to deliver 1 A cm−2) for 1000 h. Overall, the synergistic interplay of the Se-RuOx catalyst with rapid electron diffusion and recovery capabilities provides prospective insight into rationally creating LOM-OVSM catalysts that notably enhance both OER activity and stability.
Methods
Chemicals and materials
All chemicals were used as received. Ruthenium(III) chloride (RuCl3, ≥99.9%), selenium (Se, ≥99%), sulfur sublimation (S, ≥99%), sodium monophosphate (NaH2PO2, ≥99%), Vulcan XC 72 carbon, urea (CH4N2O, ≥99%), absolute ethanol (C2H6O, ≥99.7%), Nafion117 (≈5%), and isopropanol (C3H8O, ≥99.7%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Carbon cloth (WOS 1011) was obtained from CeTech. High-purity water was acquired from the Milli-Q purification system.
Fabrication of RuO2
First, 0.5 mmol of RuCl3 was dissolved in 4 mL of absolute ethanol, and then the solution was mixed well by stirring. The product was collected by heating to 600 °C using a muffle furnace at a ramp rate of 5 °C/min for 120 min and then cooling to room temperature at a ramp rate of 5 °C/min.
Fabrication of R-RuOx (R = P, S, N, Se) and Se-RuOx-y (y = 1, 2, 4, 6)
The synthesis of R-RuOx followed the same procedure as the previously described synthesis method, with an additional 0.02 mmol of Se (or S, CH4N2O, NaH2PO2) in anhydrous ethanol.
The synthesis of Se-RuOx-y (y = 1, 2, 4, or 6) followed the same procedure as the synthesis method, with different Se powder contents (0, 0.01, 0.02, 0.04 and 0.06 mmol, respectively) in anhydrous ethanol.
Characterizations
X-ray diffraction (XRD) patterns were collected on an Empyrean PANalytical diffractometer with Cu-Kα radiation (40 kV, 40 mA, λ = 1.5418 Å) at a scanning rate of 5° min−1. JEM-2100F was used to acquire the TEM, HAADF-STEM, and EDS mapping images. A Thermo Scientific K-Alpha+ instrument was used to collect X-ray photoelectron spectrometry (XPS) information. The calibration peak data were based on the C 1 s peak at 284.8 eV. All vibrating sample magnetometer (VSM) data were collected via a Quantum Design-PPMS9. The magnetic hysteresis (M-H) curve was measured at ±3 T magnetic field strength at room temperature, and the temperature-dependent magnetization (M-T) curve was measured at H = 1 K Oe magnetic field strength at temperatures ranging from 2–300 K. Electron paramagnetic resonance (EPR) was performed on a Bruker A300 at 77k temperature to obtain oxygen vacancy correlation information. The g value can be calculated from the original data via the following equation: g = hν/βH, where h is the Plank constant with a value of 4.135 × 10−15 eV s, ν is the microwave frequency of the X-band spectrometer, β is the electron Bohr magneton with a value of 5.788×10−5 eV T−1, and H is the applied magnetic field. For X-ray absorption spectroscopy (XAS) analysis, test samples were prepared on titanium foil and subsequently brushed off to collect powders. The resultant powders were evenly applied onto a 10 cm long and 1 cm wide piece of plastic, radiation-resistant Scotch tape (commonly known as Kapton tape, often referred to as the duct tape of synchrotrons). In order to obtain a compact particle distribution on the Scotch tape without any pinholes, following the removal of larger particles by shaking, the sample tape was divided into 10 smaller segments. These segments were then layered one on top of the other onto a separate piece of Scotch tape and subsequently encapsulated with an additional layer of Scotch tape. The Ru K-edge X-ray absorption spectra (XAS) were collected at the 1W1B beamline of the Beijing Synchrotron Radiation Facility in the People’s Republic of China, which operates at a current of 200 mA and an energy of 2.5 GeV. Ru foil served as the calibration standard, with all specimen analyses conducted using transmission-mode spectroscopy. The XAFS data were processed utilizing the Demeter software package. Normalization of the spectra was initially carried out using Athena, followed by shell fitting analyses performed with Artemis. The χ(k) function was subjected to Fourier transformation (FT) employing k3 weighting, and all fitting procedures were conducted in R space.
In situ Raman
In situ Raman spectroscopic measurements were conducted through a confocal microscopy system (DXR platform) interfaced with an electrochemical flow cell. Experimental configurations utilized a 10× magnification objective and 2.0 mW laser excitation energy. The working electrode was Se-RuOx/Au, the counter electrode was a platinum wire, and the reference electrode was a saturated calomel electrode. The flow rate of the electrolyte was 5 mL min−1 to remove the oxygen produced in the reaction process. After applying a constant potential for 20 s, the sample information was gathered, and the in situ Raman spectra of each working electrode were captured three times. Within the voltage range of 0.9–2.0 V vs. SCE, a variation in the Raman spectra became evident.
In situ ATR-SEIRAS
In situ FTIR experiments were carried out with a custom-designed cell. The catalysts were deposited on a GCE positioned at the center of the cell, and a Pt wire as well as a saturated calomel electrode were used as the counter and reference electrodes, respectively. In order to avoid electrolyte evaporation that could damage the microscope lens, a BaF2 window was positioned above the cell. The OER was conducted with linearly increasing potentials from open-circuit potential (OCP) to 1.85 V vs. RHE at fixed polarization, during which synchrotron radiation Fourier-transform infrared (SR-FTIR) spectral data were collected through 100-scan signal accumulation with 1 cm⁻¹ spectral resolution. First, the background spectrum was recorded without applying any voltage. During data processing, the background spectrum was subtracted from the infrared spectrum of the sample.
Electrochemical measurements
The OER catalytic performance was evaluated in a three-electrode system on a CHI760e electrochemical workstation at 25 °C. Electrolyte solutions are generally prepared fresh before use. Three milligrams of R-RuOx were dispersed in 600 μL of 0.1% Nafion solution (diluted with isopropanol) and ultrasonicated for 20 min to disperse uniformly, after which 21 μL of the resulting suspension was deposited onto a glassy carbon (GC, d = 0.5 cm) electrode. All control samples exhibited comparable mass loading. Briefly, the synthesized catalysts were used as working electrodes, while a carbon rod and a saturated calomel electrode (SCE, Hg/Hg2Cl2) functioning as the counter and reference electrode, respectively. Linear sweep voltammetry (LSV) curves were conducted in a 0.5 M H2SO4 solution (pH≈0.3) at a scan rate of 5 mV/s with 85% inner resistance (iR) compensation. E (vs. RHE) = E (vs. SCE) + 0.059*pH + 0.241. All the chronopotentiometric stability tests were evaluated in 0.5 M H2SO4 using a standard three-electrode system with a carbon cloth electrode. All OER performances were evaluated in 0.5 M HSO4 with iR correction (85% iR-correction set via the instrument). For Tafel analysis, the potential corresponding to a current density near 10 mA cm⁻² was chosen. The Faradaic efficiency was determined by gathering O2 and H2 at a constant current of 0.1 A (25 °C, 1 atm). Electrochemical impedance spectroscopy (EIS) was conducted across a frequency spectrum extending from 100 kHz to 0.01 Hz with applied voltages set at 1.25 V, 1.3 V, 1.35 V, 1.4 V, 1.45 V, 1.5 V, and 1.55 V. Electrochemically active surface areas (ECSAs) were determined using cyclic voltammetry (CV) within a potential range of 1.0–1.1 V versus SCE, at scan rates of 10, 20, 30, 40, and 50 mV s−1. The ECSA was calculated based on the current density measured at 1.05 V versus SCE. The electrical double-layer capacitance (Cdl) was estimated using the formula Cdl = (ja-jc)/2ν, where ja and jc represent the anodic and cathodic current densities, respectively, and ν denotes the scan rate. Consequently, Cdl corresponds to the slope of the linear relationship between (ja-jc)/2 and the scan rate. The ECSA was then derived using the equation ECSA = Cdl/Cs (Cs = 0.040 mF cm−2).
PEMWE electrolyser
Membrane electrode assemblies (MEAs) were fabricated utilizing a Nafion®211 polymer membrane (active area of 1 cm2, film thickness: 25.4 μm). Se-RuOx (2 mg cm−2) and commercial Pt/C (1 mg cm−2) catalysts were used as the anode and cathode, respectively. Electrochemical evaluation was carried out in a single-cell PEM water electrolyzer (PEMWE) configuration. Porous titanium felt layers served as both anode and cathode gas diffusion media. For durability analysis, stability measurements were executed using a Keithley 2461 Source Meter instrumentation platform.
DFT calculation details
DFT calculations were carried out using the VASP, employing the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) and the projector augmented wave (PAW) method. The interaction between core and valence electrons was described using PAW approach57. The crystal structure of RuO2 is shown in Fig. S52, and DFT-optimized equilibrium lattice constants are a = 4.492, b = 4.492, c = 3.107 Å, which is inconsistent with previous reports58. To discuss the impact on the doping sites of Se atom, we substituted three Se atoms along the x, y, and z directions to simulate Se-RuOx. Additionally, we substituted 1, 2, and 3 Se atoms at the Ru-Ru sites along the z-direction to verify the effect of different doping ratio on Ru. Periodically repeated three-layer slab is modeled for RuO2 (110) as the active surfaces to investigate the OER performance. The bottom layer of the slab was fixed during structural optimization, while the top two layers were relaxed, using a 4×4×1 Monkhorst–Pack k-point mesh for Brillouin zone sampling. A plane-wave basis set with a kinetic energy cutoff of 450 eV was employed to expand the valence electron wavefunctions, and a Gaussian smearing width of 0.05 eV was utilized during structural optimization. The electronic self-consistent field (SCF) calculations were considered converged when the total energy change was below 10⁻⁵ eV, and atomic structures were optimized until the residual forces were smaller than 0.02 eV/Å. For Ru atoms, an effective Hubbard parameter (U–J) of 4.2 eV was applied.
The OER process via LOM-mediated process can be represented by the following stepwise reactions59,60:
As a result, the Gibbs free energy variations were determined as follows:
The free energy of O₂ was derived from the reference reaction: H₂O(l) → H₂(g) + O₂(g).
The overpotential (η), a key descriptor of OER activity, can be directly obtained based on the following proposed OVSM pathway for the oxygen evolution reaction59,60:
where Vo-M* denotes the active site involved in the reaction; G₁, ΔG₂, ΔG₃, and ΔG₄ correspond to the free energy changes for each individual step; and
and η = max (ΔG1, ΔG2, ΔG3, ΔG4) – 1.23.
Data availability
The data that support the conclusions of this study are available within the paper and in the Supplementary Information, and Supplementary Data 1 for the atomic coordinates of the optimized unit cell. Source data are provided with this paper.
References
Lina, C. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Yang, S. et al. The mechanism of water oxidation using transition metal-based heterogeneous electrocatalysts. Chem. Soc. Rev. 53, 5593–5625 (2024).
Zhao, C. X. et al. Recent advances of noble-metal-free bifunctional oxygen reduction and evolution electrocatalysts. Chem. Soc. Rev. 50, 7745–7778 (2021).
Wang, X. et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 611, 702–708 (2022).
Mou, T. et al. Rationalizing acidic oxygen evolution reaction over iro2: essential role of hydronium cation. Angew. Chem. Int. Ed. 48, e202409526 (2024).
Liu, J. et al. Single-atom Co dispersed on polyoxometalate derivatives confined in bamboo-like carbon nanotubes enabling efficient dual-site lattice oxygen mediated oxygen evolution electrocatalysis for acidic water electrolyzers. Energy Environ. Sci. 17, 3088–3098 (2024).
Ram, R. et al. Water-hydroxide trapping in cobalt tungstate for proton exchange membrane water electrolysis. Science 384, 1373–1380 (2024).
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science 384, 666–670 (2024).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558–573 (2023).
Zhao, G. et al. Metallic Ru-Ru Interaction in Ruthenium Oxide Enabling Durable Proton Exchange Membrane Water Electrolysis. Adv. Mater. 36, e2404213 (2024).
Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Yan, H. et al. Ultrathin carbon coating and defect engineering promote ruo2 as an efficient catalyst for acidic oxygen evolution reaction with super-high durability. Adv. Energy Mater. 13, 2300152 (2023).
Ping, X. et al. Locking the lattice oxygen in RuO2 to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Chen, F.-Y. et al. Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design. Joule 5, 1704–1731 (2021).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304–313 (2019).
Cui, X. et al. Robust interface ru centers for high-performance acidic oxyGEN EVOLUtion. Adv. Mater. 32, e1908126 (2020).
Gou, W. et al. Oxygen spillover from RuO2 to MoO3 enhances the activity and durability of RuO2 for acidic oxygen evolution. Energy Environ. Sci. 17, 6755–6765 (2024).
Du, K. et al. Interface engineering breaks both stability and activity limits of RuO2 for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Chang, J. et al. Oxygen Radical Coupling on Short-Range Ordered Ru Atom Arrays Enables Exceptional Activity and Stability for Acidic Water Oxidation. J. Am. Chem. Soc. 146, 12958–12968 (2024).
Fan, R. Y. et al. The promising seesaw relationship between activity and stability of ru-based electrocatalysts for acid oxygen evolution and proton exchange membrane water electrolysis. Small 20, e2304636 (2024).
Wang, C. et al. Identification of the origin for reconstructed active sites on oxyhydroxide for oxygen evolution reaction. Adv. Mater. 35, e2209307 (2023).
Miao, L. et al. Computational chemistry for water-splitting electrocatalysis. Chem. Soc. Rev. 53, 2771–2807 (2024).
Lei, X. et al. Unraveling the oxygen vacancy site mechanism of a self-assembly hybrid catalyst for efficient alkaline water oxidation. ACS Catal. 14, 4523–4535 (2024).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Sun, Y. et al. Nitrogen-doped cobalt diselenide with cubic phase maintained for enhanced alkaline hydrogen evolution. Angew. Chem. Int. Ed. 60, 21575–21582 (2021).
Sun, N. et al. Augmented Electrochemical Oxygen Evolution by d–p Orbital Electron Coupling. Adv. Mater. 36, 2404772 (2024).
Yao, N. et al. Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem 9, 1882–1896 (2023).
Wang, J. et al. Single-site Pt-doped RuO2 hollow nanospheres with interstitial C for high-performance acidic overall water splitting. Sci. Adv. 8, eabl9271 (2022).
Chen, D. et al. Bicontinuous RuO2 nanoreactors for acidic water oxidation. Nat. Commun. 15, 3928 (2024).
Wang, K. et al. Highly active ruthenium sites stabilized by modulating electron-feeding for sustainable acidic oxygen-evolution electrocatalysis. Energy Environ. Sci. 15, 2356–2365 (2022).
Lee, K. et al. Modulating the valence electronic structure using earth-abundant aluminum for high-performance acidic oxygen evolution reaction. Chem 9, 3600–3612 (2023).
Wang, Y. et al. Intercalant-induced V t2g orbital occupation in vanadium oxide cathode toward fast-charging aqueous zinc-ion batteries. Proc. Natl Acad. Sci. Usa. 120, e2217208120 (2023).
Zhang, H. et al. Oxygen vacancies unfold the catalytic potential of nife-layered double hydroxides by promoting their electronic transport for oxygen evolution reaction. ACS Catal. 13, 6000–6012 (2023).
Xu, Y. et al. Strain-modulated Ru-O Covalency in Ru-Sn Oxide Enabling Efficient and Stable Water Oxidation in Acidic Solution. Angew. Chem. Int. Ed. 63, e202316029 (2024).
Su, R. et al. Utilizing the oxygen-atom trapping effect of Co3O4 with oxygen vacancies to promote chlorite activation for water decontamination. Proc. Natl Acad. Sci. Usa. 121, e2319427121 (2024).
Shen, R. et al. Engineering bimodal oxygen vacancies and pt to boost the activity toward water dissociation. Small 18, e2105588 (2022).
Chen, Y. et al. Metastabilizing the ruthenium clusters by interfacial oxygen vacancies for boosted water splitting electrocatalysis. Adv. Energy Mater. 14, 2400059 (2024).
Shen, G. et al. Regulating the Spin State of FeIII by atomically anchoring on ultrathin titanium dioxide for efficient oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 59, 2313–2317 (2020).
SUNTIVICH, J. et al. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334, 1383–1385 (2011).
Zhang, N. et al. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Zhu, J. et al. Ultrahigh Stable Methanol Oxidation Enabled by a High Hydroxyl Concentration on Pt Clusters/MXene Interfaces. J. Am. Chem. Soc. 144, 15529–15538 (2022).
Jia, C. et al. Shifting oxygen evolution reaction pathway via activating lattice oxygen in layered perovskite oxide. Adv. Funct. Mater. 33, 2301981 (2023).
Zhang, J. et al. Modulation for RuO2/TiO2 via Simple Synthesis to Enhance the Acidic Oxygen Evolution Reaction. ACS Sustain. Chem. Eng. 11, 9489–9497 (2023).
Yan, S. et al. Highly active and stable alkaline hydrogen evolution electrocatalyst based on ir-incorporated partially oxidized ru aerogel under industrial-level current density. Adv. Sci. 11, e2307061 (2024).
Hodnik, N. et al. New insights into corrosion of ruthenium and ruthenium oxide nanoparticles in acidic media. J. Phys. Chem. C. 119, 10140–10147 (2015).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Su, Z. et al. Probing the actual role and activity of oxygen vacancies in toluene catalytic oxidation: evidence from in situ XPS/NEXAFS and DFT +UCalculation. ACS Catal. 13, 3444–3455 (2023).
Ye, K. et al. An overview of advanced methods for the characterization of oxygen vacancies in materials. TrAC, Trends Anal. Chem. 116, 102–108 (2019).
Du, Y. et al. 3D hierarchical fireproof gel polymer electrolyte towards high-performance and comprehensive safety lithium-ion batteries. Chem. Eng. J. 476, 146605 (2023).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659–23669 (2023).
Xu, Z. et al. Surface reconstruction facilitated by fluorine migration and bimetallic center in nico bimetallic fluoride toward oxygen evolution reaction. Adv. Sci. 11, e2306758 (2024).
Jia, H. et al. Stabilizing atomic Ru species in conjugated sp2 carbon-linked covalent organic framework for acidic water oxidation. Nat. Commun. 15, 5419 (2024).
Jia, H. et al. Understanding the Role of Spin State in Cobalt Oxyhydroxides for Water Oxidation. Angew. Chem. Int. Ed. 47, e202408005 (2024).
Zhong, H. et al. Key role of eg* band broadening in nickel-based oxyhydroxides on coupled oxygen evolution mechanism. Nat. Commun. 14, 7488 (2023).
Tang, J. et al. Ruthenium single-atom modulated protonated iridium oxide for acidic water oxidation in proton exchange membrane electrolysers. Adv. Mater. 36, 2407394 (2024).
Kresse, G. et al. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Haines, J. et al. Neutron diffraction study of the ambient-pressure, rutile-type and the high-pressure, CaCl2-type phases of ruthenium dioxide. Acta Crystallogr. Sect. B 53, 880–884 (1997).
Lu, Q. et al. Breaking the activity-stability trade-off of ruo2 via metallic ru bilateral regulation for acidic oxygen evolution reaction. Angew. Chem. Int. Ed. 64, e202503733 (2025).
Zhang, D. et al. Construction of Zn-doped RuO2 nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22309137, 22279095), the Department of Science and Technology of Hubei Province (2024CSA076), and the Hubei Key Laboratory for New Textile Materials and Applications Research (FZXCL202206). We acknowledge the great help from Prof. Weilin Xu and coworkers at Wuhan Textile University for helpful measurements and discussion. We thank the Analytical and Testing Center of Wuhan Textile University for the XRD and ICP tests. The computation is completed in the HPC Platform of Wuhan Textile University. We are grateful to Professor Wan Renzhuo and Professor Yang Fan for their support in the field of computation.
Author information
Authors and Affiliations
Contributions
N.Y. and X.W. conceived and designed the project and cowrote the paper. W.P. conducted the experiments, synthesized and characterized the samples and analysed the data. N.Y. performed the theoretical modelling. Z.L. and S.H. helped with the synchrotron experiments. H.B. and W.X. provided input during the writing process. All the authors discussed the results and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Pi, W., Li, Z. et al. Orbital-level band gap engineering of RuO2 for enhanced acidic water oxidation. Nat Commun 16, 4845 (2025). https://doi.org/10.1038/s41467-025-60083-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60083-y
