
Abstract
Human recombination maps are a valuable resource for association and linkage studies and crucial for many inferences of population history and natural selection. Existing maps1,2,3,4,5 are based solely on cross-over (CO) recombination, omitting non-cross-overs (NCOs)—the more common form of recombination6—owing to the difficulty in detecting them. Using whole-genome sequence data in families, we estimate the number of NCOs transmitted from parent to offspring and derive complete, sex-specific recombination maps including both NCOs and COs. Mothers have fewer but longer NCOs than fathers, and oocytes accumulate NCOs in a non-regulated fashion with maternal age. Recombination, primarily NCO, is responsible for 1.8% (95% confidence interval: 1.3–2.3) and 11.3% (95% confidence interval: 9.0–13.6) of paternal and maternal de novo mutations, respectively, and may drive the increase in de novo mutations with maternal age. NCOs are substantially more prominent than COs in centromeres, possibly to avoid large-scale genomic changes that may cause aneuploidy. Our results demonstrate that NCOs highlight to a much greater extent than COs the differences in the meiotic process between the sexes, in which maternal NCOs may reflect the safeguarding of oocytes from infancy until ovulation.
Similar content being viewed by others

Main
Sexually reproducing organisms make gametes through meiosis7, in which homologous chromosomes recombine, ensuring proper segregation of chromosomes and mixing the genetic material that is passed on to the offspring. Human genetic diversity and our ability to evolve—and thereby adapt—is generated by de novo mutations (DNMs) and meiotic recombination. DNMs are known to occur in part as a result of recombination1,8,9,10,11, but, as NCOs are under-reported, the extent to which recombination contributes to DNMs in the offspring remains unknown. Meiosis is initiated by duplication of the genetic material and subsequent formation of double-strand breaks (DSBs)12, occurring mainly in hotspots defined by histone methyltransferase PRDM9 (ref. 13). PRDM9 recruits SPO11, which, together with TOP6BL14, mediates DNA cleavage, resulting in DSBs15. The 5′ ends of double-stranded DNA at a DSB are resected (Fig. 1a), leaving an overhang of 3′ single-stranded DNA (ssDNA) branches16. The homologous chromosome is subsequently invaded by one or both 3′ branches for repair of the DSB through DNA resynthesis, resulting in a recombination, either a cross-over (CO) or a non-cross-over (NCO)6,17.

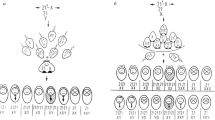
a, Schematic view of NCO and CO resolution. A DSB is induced on one chromosome (red) and the 5′ strands near the DSB are resected. The 3′ strands invade the homologous chromosome (blue), and DNA is synthesized (dotted lines) to bridge the DSB. When only one strand invades, the synthesis-dependent strand annealing (SDSA) pathway is used, leading to NCOs. When both strands invade, a double Holliday junction (dHJ) is generated; this is the primary source of COs. b, Schematic view of recombination events. The points denote MPPs in a meiosis, with the colour indicating the grandparental origin of each MPP. Short haplotype segments are gene conversion candidates flanked by background haplotypes forming (i) a simple oNCO with a single converted segment or (ii) a complex oNCO with alternating gene-converted and non-gene-converted segments if background haplotypes are of the same grandparental origin, or otherwise (iii) a CO with associated gene conversions. c, Schematic overview of the NCOurd process and subsequent analysis. (i) oNCOs are specified by a set of gene-converted MPPs (red) and the surrounding background haplotype MPPs (blue). Our previously described method23 (NCOurd) derives length distributions for NCOs from the oNCOs. These are used to compute the numbers of NCOs per offspring or region. NCOs per offspring allow us to explore sex differences and age dependence of the meiotic process, as well as interactions with DNMs estimated from (ii) DNMs found near oNCOs. NCOs per region are used to compute the number of NCOs throughout the genome to create maps of NCO activity and DSB resolution.
For DSBs repaired as NCOs, the resected region around the DSB is partially repaired using the homologous chromosome as a template. Thus, NCOs manifest as short transitions between haplotypes of different grandparental origin, distinguished only at markers that are heterozygous in the transmitting parent (Fig. 1b). NCOs that have incorporated a donor sequence that includes a heterozygous marker in the parent will contain heteroduplex DNA16 with mismatched base-pairings that must be resolved. Some of these are resolved to match the original haplotype, leaving no evidence of the NCO in the offspring. However, when they are resolved to match the donor sequence, a haplotype transition will be detectable; this is referred to as a gene conversion18. Meiotic DSBs may be repaired using the sister chromatid as the template, in which case neither COs nor NCOs would be detectable owing to the near-perfect sequence identity between the sister chromatids. Sister chromatid repair is suppressed in meiosis and is therefore rare6. We focus on DSBs that were repaired using the homologous chromosome in meiocytes that resulted in live births.
The meiotic process differs markedly between males and females19. Spermatogonia undergo continual mitosis from puberty throughout life, whereas meiosis is initiated in spermatocytes only a few weeks before haploid sperm cells are fully formed. By contrast, females are born with a limited supply of oocytes with the meiotic process suspended in prophase I, only to be completed at ovulation and fertilization decades later20. Oocytes are thus exposed for much longer to possible exogenous agents that may adversely affect DNA integrity21, and some of their NCOs may be due to repair of DSBs sustained and accumulated over the years.
We have derived complete sex-specific recombination maps, including both NCOs and COs, thereby completing the high-resolution mapping of sex-specific human recombination, a task we started more than 20 years ago2. We identified NCOs by looking for gene conversions (Supplementary Note 1.1) transmitted to offspring in 5,420 trios in 2,132 Icelandic nuclear families in which both parents and at least two children have undergone whole-genome sequencing (WGS)22 (Supplementary Table 1). We estimated the length distribution of NCOs23 and their average number per meiosis and constructed genetic maps of NCOs (Fig. 1c). By identifying DNMs near NCOs in the same individuals, we derived sex-specific mutational spectra of DNMs resulting from NCOs and estimated the overall contribution of recombination to mutagenesis.
Gene conversions and observed NCOs
We restricted our analysis to autosomal variants with frequencies above 0.5% (Supplementary Note 1.4), resulting in 8,893,878 sequence variants, 8,270,254 single-nucleotide polymorphisms (SNPs) and 623,624 insertions and/or deletions (indels) (Supplementary Table 2). Gene conversions could only be determined at variants for which the transmitting parent was heterozygous, and we required the other parent to be homozygous (Supplementary Note 2), giving totals of 4,229,340,533 and 4,288,524,590 informative marker–proband pairs (MPPs) for paternal and maternal meiosis, respectively (Extended Data Table 1). When the genotypes of parent and offspring were phased, and the parent of origin had been determined, contiguous haplotype segments of a given grandparental origin could be identified in offspring (Supplementary Note 3). Telomeric haplotype segments and haplotype segments longer than 100 kb were considered to represent background, consistent with reciprocal recombination, whereas haplotype segments shorter than 100 kb were considered to be gene conversion candidates24 (Fig. 1b(i)). Consecutive gene conversion candidates flanked by two background haplotypes of the same grandparental origin were considered to be observed NCOs (oNCOs); note that oNCOs may be complex, containing both gene-converted and non-gene-converted MPPs (Fig. 1b(ii)). When the grandparental origins of the flanking background haplotypes differ, the result is a complex CO1 (Fig. 1b(iii)).
We identified 17,109 paternal and 45,653 maternal gene-converted MPPs—an increase of more than 30-fold compared with the largest family-based study so far24 (Extended Data Table 1). Gene conversion rates were consistent with earlier estimates24,25. The total numbers of oNCOs were 12,948 and 15,712, with averages of 2.39 (95% confidence interval (CI): 2.33–2.44) and 2.90 (95% CI: 2.83–2.97) oNCOs per offspring for paternal and maternal meioses, respectively, and the mean numbers of converted MPPs per oNCO were 1.32 (95% CI: 1.26–1.40) and 2.91 (95% CI: 2.79–3.03) for paternal and maternal meiosis, respectively. A larger study26 of gene conversions identified from identity-by-descent segments was published concurrently with this research. We observed a GC bias in gene-converted SNPs (Extended Data Table 1), with strong (C or G) alleles more likely to be transmitted than weak (A or T) ones, consistent with earlier studies24,25,27,28. We noted a slightly higher GC bias (P = 0.002, bootstrap test) for maternal transversion: 67.1% (95% CI: 65.8–68.5) versus 65.1% (95% CI: 64.2–65.9) for transitions, with 50% indicating no bias. For indels, we replicated the insertion bias reported in refs. 11,24 (the longer allele was retained more frequently than the shorter one). This bias was restricted to maternal meiosis, for which it was 62.5% (95% CI: 60.4–64.4, P = 1.7 × 10−36, binomial test).
Mothers transmit fewer and longer NCOs
From oNCOs identified for each proband, we estimated, separately for fathers and mothers, the length distribution of all NCOs, including those that were not observable owing to the absence of gene-converted MPPs. We modelled the length distributions as mixtures of several components (Supplementary Note 4) using our previously described method23. Very short NCOs (less than 10 bp) would not be accurately identified from parent offspring transmissions; however, our data suggest that these are rare (Extended Data Fig. 1). The length distributions consisted of multiple components (Supplementary Tables 3 and 4), which we grouped into short components (less than 1 kb) and extended components. Short NCOs were, on average, 123 bp (95% CI: 94–135) and 102 bp (95% CI: 71–125) for paternal and maternal transmissions, respectively (Extended Data Table 1). The size difference between the sexes was not significant (P = 0.272, bootstrap test), and the results were comparable with earlier NCO length estimates of 55–290 bp in humans29, 30 bp in baboons30 and 155 bp in rhesus macaques31, and, in mice32, 86 bp (range 23–148) in oocytes and 68 bp (range 15–124) in spermatocytes. All these estimates are shorter than the size of resected regions around DSBs, which have been estimated to be approximately 1,400 bp in human testis10, 1,640 bp in yeast33 and 2,200 bp in mice34, all with wide variability. Extended NCOs are, on average, 7.2 kb (95% CI: 1.8–11.8) and 9.1 kb (95% CI: 6.8–10.8), for paternal and maternal transmissions, respectively. Extended NCOs represent 1.14% (95% CI: 0.62–5.88) of paternal NCOs but are more common in mothers (6.89% (95% CI: 5.10–9.93)) and appear to be generated from a different process than the paternal ones, with stronger allele selection biases (Extended Data Table 1 and Supplementary Table 5).
On the basis of the inferred length distribution, we estimated that there would be, on average, 105.0 (95% CI: 95.9–125.0) and 81.6 (95% CI: 66.7–103.1) NCOs per offspring for paternal and maternal meiosis, respectively (Fig. 1c and Supplementary Note 4.3), indicating that the vast majority of NCOs are unobserved. We estimated that there would be more NCOs in paternal meiosis (P = 0.042, bootstrap test), in contrast to the greater number of COs in maternal meiosis1 (approximately 42 versus approximately 26 for paternal meiosis). Estimating DSBs by combining our estimates for NCOs and COs (Supplementary Note 4.4), we found that the average number of DSBs did not significantly differ between the sexes (P = 0.120, bootstrap test), with the average numbers of DSBs per meiocyte estimated to be 474 (95% CI: 438–554) and 410 (95% CI: 350–496) for paternal and maternal meiosis, respectively. Cytological measurements of early recombination intermediates in human meiocytes give estimates of approximately 400 DSBs in spermatocytes35 and 370 DSBs in oocytes36.
NCOs and COs were positively correlated for both paternal and maternal meiosis (r = 0.058, P = 2.0 × 10−5 paternal; r = 0.100, P = 1.6 × 10−13 maternal; Supplementary Note 4.5). This is consistent with an increase in DSBs leading to an increase in both NCOs and COs, in both fathers and mothers, and indicates that an increase in COs does not come at the cost of fewer NCOs. These results are also consistent with a model in which chromosomal axis lengths vary among germ cells, whereas the distance between DSB events varies less37. The relatively modest correlation may be explained in part by CO homeostasis17,38.
Genic DSBs favour NCO resolution
We constructed sex-specific maps of human NCO recombination on a grid of overlapping 3-Mb windows at intervals of 1 Mb (Supplementary Note 5), achieving resolution on par with that of early CO maps2,4. Human recombination maps1,2,3,4,5 have only accounted for CO recombination, but combining our NCO maps with existing CO maps1 allowed us to explore all recombination as well as variations in DSB resolution through differences in the NCO and CO maps. We display the sex-specific NCO maps for chromosome 19 in Fig. 2a and the full set of recombination maps in Supplementary Fig. 1.

a, NCO maps for chromosome (chr.) 19. b, ΔDSB measure for chr. 19. Cytobands are shown below the graphs, with the centromere indicated in red, gneg bands in white, all gpos bands in grey, and gvar and stalk bands in blue. c,d, Average values of NCO (c) and ΔDSB (d) recombination maps near telomeres, with the NCO data normalized to the autosomal average. Error bars show 95% confidence intervals computed by bootstrapping 1,000 samples on the basis of map data for the 22 autosomes. e,f, As in c (e) and d (f), but for map values near centromeres. Dashed lines represent genome-wide averages. g,h, Results for per-offspring NCO count (g) and ΔDSB (h) of maternal meioses versus maternal age. Offspring are grouped by maternal age in bins of size 2 years; the points show group averages, omitting bins with fewer than 25 offspring. Error bars show 95% confidence intervals computed by bootstrapping 1,000 samples from the 5,240 probands. Green lines show linear regression results using the inverse of the size of the confidence intervals as weight. P values for regression results were based on Student’s t-distribution.
Large-scale features of the CO map, such as the elevation of paternal rates near telomeres, are mirrored in the NCO map, and we found that these maps were highly correlated—the genome-wide correlations were 0.68 (95% CI: 0.65–0.71) and 0.36 (95% CI: 0.32–0.40) for paternal and maternal maps, respectively. There was significant positive correlation between the paternal CO and NCO maps for all autosomes, whereas the maternal maps were significantly positively correlated for all autosomes except for 6, 16, 17, 21 and 22 (Supplementary Table 6).
Highlighting the shared aetiology of NCOs and COs, locations of oNCOs were highly correlated with sex-specific CO recombination hotspots1 (Extended Data Table 2 and Supplementary Note 6), where their frequency was increased 22.4-fold and 13.7-fold for fathers and mothers, respectively. Furthermore, the hotspot usage of oNCOs was associated with a PRDM9 sequence variant (rs2973614, major allele frequency: 3.2%; effect = −0.6 s.d., P = 1.1 × 10−21; Supplementary Note 7) that was also strongly associated with hotspot usage of COs1,13 (effect = −1.7 s.d., P = 4.3 × 10−2,382). NCOs were also highly enriched in regions of annotated DSB activity (Extended Data Table 2), with the highest enrichments found in DNA meiotic recombinase 1 (DMC1) hotspots10,11 (42.4-fold and 19.3-fold, for paternal and maternal NCOs, respectively). NCOs also colocalized with binding sites of PRDM9 (ref. 39) (Supplementary Note 8), and this binding was biased with respect to the centre position of oNCOs; the centre of the PRDM9 binding motif was, on average, 36 bp (95% CI: 26.5–41.0 bp) downstream of the centre position of the oNCO (Supplementary Table 7), consistent with PRDM9 inducing DSBs preferentially upstream of its binding site.
There was an excess of NCOs in exons and transcribed regions40 (Extended Data Table 2); the enrichments in transcribed regions were 1.52 (95% CI: 1.38–1.67) and 1.26 (95% CI: 1.17–1.36) for paternal and maternal meiosis, respectively. These results run counter to the well-documented depletion of COs in such regions1,41 (Extended Data Table 2). Transcription is marked by H3K36 trimethylation40—known to be deposited by the DSB-associated PRDM9 (ref. 42) —which has been potentially implicated in regulating DSB pathway choice43, possibly explaining why DSB resolution is NCO-enriched in transcribed regions.
Pericentromeric DSBs are resolved as NCOs
The NCO/CO ratio is an indicator of the propensity of DSBs to be resolved as NCOs rather than COs; here, we estimated this ratio to be 7.84 (95% CI: 7.24–9.49) per meiocyte for paternal meiosis and 3.91 (95% CI: 3.15–4.93) per meiocyte for maternal meiosis. These estimates are concordant with prior estimates29,44 and with Housworth–Stahl modelling of CO interference (Supplementary Note 9). We analysed localized variations in the NCO/CO ratio through a normalized NCO/CO difference, ΔDSB, which measures how DSB resolution deviates locally from the genome-wide average of the NCO/CO ratio (Fig. 2b). Values of ΔDSB ranged from −1 (only COs) to 1 (only NCOs). Here, ΔDSB > 0 indicates that DSBs are resolved as NCOs at a higher rate than the genome-wide average, referred to as NCO-enriched DSB resolution, whereas ΔDSB < 0 indicates CO-enriched DSB resolution.
Paternal DSB rates were highly elevated in the 10-Mb region closest to the telomere, with both NCOs and COs contributing to this (Fig. 2c and Extended Data Fig. 2). Paternal DSB resolution was NCO-enriched in the 1-Mb interval closest to the telomere; however, in the range 3–10 Mb from the telomere, the resolution was CO-enriched45. Maternal DSB rates, on the other hand, were only mildly elevated in the 10-Mb region closest to the telomere; this was driven by elevation of NCOs and NCO-enriched resolution up to 5 Mb from the telomere. Further away (up to 10 Mb), maternal CO rates were also elevated, and the DSB resolution was balanced between COs and NCOs (Fig. 2d and Extended Data Fig. 2).
Although COs are known to be suppressed near centromeres46,47,48, our results (Fig. 2e) demonstrate that the processes that govern that suppression do not affect NCOs49; consequently, we observed NCO-enriched DSB resolution near centromeres (Fig. 2f and Supplementary Fig. 1). This was especially evident for paternal meiosis, for which the average ΔDSB was about 0.85 within 1 Mb of the centromere (indicating that almost all DSBs are resolved as NCOs) and remained significantly positive up to 10 Mb away from the centromere. For maternal meiosis, this suppression is less effective, as the average ΔDSB was positive only within 1 Mb of the centromere.
Recombination map values and DSB resolution as functions of both GC content and replication timing are displayed in Extended Data Figs. 3 and 4, respectively.
DNMs are enriched near oNCOs
We have previously described genomic regions with high NCO rate and maternal C>G DNM rate (C>G-enriched regions; CGER9). However, as relatively few NCOs were detected per individual and oNCO datasets have been limited in size, we did not detect co-occurrence of DNMs and NCOs in the same meiosis. Here, we expanded this and explored colocalization of oNCOs with 382,566 DNMs identified in the probands of our study (Supplementary Note 10). For each DNM, we measured the distance to the centre of the nearest oNCO and found that within 1 kb of the oNCO centres, DNM rates were elevated 142-fold (95% CI: 106–183) and 125-fold (95% CI: 66–197) for paternal and maternal meiosis, respectively. To assess whether this enrichment was due to nucleotide composition or other genomic features of NCOs, we permuted the DNMs across the probands and recalculated the enrichment per permutation. We found no significant enrichment in the permutation, indicating that DNM and oNCO co-occurrence was not due to nucleotide composition at oNCO sites (Supplementary Table 8). The DNM rate elevation decreased rapidly for paternal meiosis and was mostly observable within 3 kb of the NCO, whereas significant elevation could be found up to 100 kb away from NCOs for maternal meiosis (Extended Data Table 3, Extended Data Fig. 5 and Supplementary Tables 9 and 10).
Sex difference in DNM spectra near NCOs
To highlight the differences in the mechanisms leading to DNMs that arise owing to NCOs versus other DNMs, we investigated the mutation spectra of the two groups of DNMs. Using our analysis of the extent of rate elevation, we used paternal and maternal DNMs within 3 kb and 100 kb, respectively, to represent DNMs that arose owing to NCOs. We grouped together mutations and their reverse complement (mutation class), considered C>T DNMs inside and outside the CpG context separately, and computed the frequency of each mutation class (mutation spectrum; Fig. 3a and Supplementary Table 11). We then tested whether the spectrum of DNMs near NCOs was different from the genomic background (P = 0.075, paternal; P = 1.5 × 10−87, maternal; χ2-test). The strong maternal difference could be mostly explained by a very large increase in the percentage of DNMs in the C>G mutation class near NCOs, 37.2% (95% CI: 31.6–43.2) compared with 7.55% (95% CI: 7.41–7.68) genome-wide (P = 4.65 × 10−49, Fisher’s test). This directly implicates NCOs with regional enrichment of maternal C>G DNMs in CGER. We next checked whether the mutational processes for NCO and CO were similar by comparing spectra for DNMs near NCOs and COs (Extended Data Fig. 6). We did not find any significant difference between the spectra for NCO-proximal and CO-proximal DNMs in paternal meiosis (P = 0.10, χ2-test); however, for maternal meiosis, they were different (P = 1.9 × 10−12, χ2-test).

a, Mutation spectra for phased DNMs proximal to oNCOs and genome-wide. DNMs were considered to be proximal to oNCOs if they were within 3 kb and 100 kb for paternally and maternally phased DNMs, respectively. The length of the bars indicates the mutation class fraction for the complete cohort of the study. Error bars show 95% confidence intervals computed by bootstrapping 1,000 samples; asterisks indicate mutation classes in which the NCO-proximal and genome-wide spectra were significantly different (P < 0.05, bootstrap test). b, Strand asymmetry for phased DNMs around oNCOs. L and R denote DNMs to the left and right of the oNCO centre, respectively.
NCOs and COs are products of DSB repair with single-stranded DNA intermediates, which are mutation prone50, and mutational and/or repair processes operating on specific types of nucleotide in a single-strand context would be expected to create strand asymmetry in the mutation spectra51. In line with this expectation, we found strand asymmetry of CpG>TpG DNMs near paternal COs1; the same pattern has been observed for C>N DNMs and sequence variants around DSB hotspots11,52. Here, we detected strand asymmetry in the spectrum for NCO-proximal DNMs (Fig. 3b and Extended Data Fig. 7). CpG>TpG asymmetry, a signature of spontaneous deamination of methylated cytosines53, was present in paternal meiosis (odds ratio = ∞ (95% CI: 2.51 to ∞, P = 4.7 × 10−3, Fisher’s test) but not in maternal meiosis. No other asymmetry signature was found in paternal meiosis; however, in maternal meiosis, we observed asymmetry in all C>N classes (Extended Data Fig. 7). These results, together with the long-range enrichment of C>G DNMs near maternal NCOs and the substantial accumulation of strand-coordinated C>G DNMs in ageing mothers9,54, indicate that the resected region around DSBs is larger than NCOs.
Age-related NCOs are not regulated
For maternal meioses, we found that the number of NCOs per offspring increased (P = 5.73 × 10−7, t-test) with maternal age at the birth of the proband by 20.3 (95% CI: 15.7–24.9) events per decade (Fig. 2g and Supplementary Table 12), in line with the age dependence observed for both CO1 and gene conversion rates24. No age effects were observed for paternal meiosis (P = 0.78, t-test), as with paternal COs1. Combining the maternal estimates, we found an increase in the number of DSBs per meiocyte of 82.5 (95% CI: 64–101) events per decade (Supplementary Table 12), with most new DSBs resolved as NCOs (Fig. 2h). Thus, pregnancies carried to term by 20-year-old mothers were products of meiocytes with an average of 349 (95% CI: 329–368) DSBs, whereas those of 40-year-old mothers resulted from meiocytes with an average of 514 (95% CI: 488–539) DSBs, a 50% increase. For a parental age of 20 years, maternal meiocytes contained significantly fewer DSBs (P < 2 × 10−3, bootstrap test) than paternal meiocytes, which contained on average 474 (95% CI: 438–554) DSBs, corresponding roughly to the average number of DSBs in 35-year-old mothers.
The increase in NCOs with maternal age occurred entirely outside DMC1 hotspots, with no change in the number of oNCOs inside the DMC1 hotspots (P = 0.88, t-test). Consequently, over a span of two decades from the age of 20 years, the percentage of oNCOs within DMC1 hotspots decreased from 29.0% (95% CI: 27.6–30.4) to 15.9% (95% CI: 14.0–17.8) (Supplementary Table 12). Both of these values were significantly smaller than the corresponding results for paternal oNCOs, which were 51.7% (95% CI: 50.7–52.7) and depended minimally on paternal age (effect = −1.75% per decade, P = 0.032, t-test). Maternal CO hotspots are another measure of programmed DSB activity in maternal meiosis. Here also, we saw a decrease in the percentage of oNCOs that fell within hotspots, from 32.8% (95% CI: 31.4–34.3) at the age of 20 years to 22.1% (95% CI: 20.1–24.1) at the age of 40 years (Supplementary Table 12). Thus, nearly all the age increase in NCOs took place outside CO hotspots.
These results indicate that maternal NCOs may become less tightly regulated with age, but we also found that maternal NCO rates increased disproportionally in CGER9; the proportion of oNCOs in CGER almost doubled for maternal age between 20 and 40, from 13.8% (95% CI: 12.5–15.0) to 24.6% (95% CI: 22.9–26.3). This increase was much greater than that for COs; thus, the meiotic NCO/CO ratio increased markedly in CGER, from 3.0 (95% CI: 2.2–3.7), to 8.4 (95% CI: 7.3–9.5), whereas outside CGER it increased from 3.3 (95% CI: 3.1–3.5) to 4.7 (95% CI: 4.4–5.0) for maternal age of 20 to 40 years (Supplementary Table 12).
Sex-specific contribution of DSB to DNMs
Extrapolating the increase in mutation rate to all NCOs (Supplementary Note 10), we found the average fractions of DNMs that could be attributed to NCOs to be 1.69% (95% CI: 1.22–2.15) and 10.95% (95% CI: 8.74–13.03) for paternal and maternal meiosis, respectively. Mutagenicity had already been established for COs1 (Supplementary Table 9), leading to estimates of about 0.11% and 0.38% for the CO contribution to the paternal and maternal mutation rates, respectively (Extended Data Table 3). The total contributions from both NCOs and COs (that is, from DSBs) were therefore 1.80% (95% CI: 1.29–2.31) and 11.3% (95% CI: 9.0–13.6) for paternal and maternal meiosis, respectively.
The increase in the mutation rate in the regions from 3–100 kb resulting from maternal NCOs was more pronounced (P < 0.002, bootstrap test) for NCOs inside CGER. Within CGER, we estimate that DSBs contribute 38.8% (95% CI: 31.4–46.7) of maternal DNMs. This percentage is strongly age dependent because of the age-related increase in the number of maternal DSBs. Thus, at the maternal age of 20 years the DSB contribution to the genome-wide DNM rate is 2.0% (95% CI: 0.8–3.7), increasing to 31.0% (95% CI: 18.6–44.8) at the age of 40 years. Within CGER, the corresponding contribution percentages are about 8.2% (95% CI: 1.9–15.8) and 80.4% (95% CI: 44.0–122.2). Thus, age-related NCOs within CGER are the main contributors to increased DNM rates in older mothers.
Discussion
We present, for both sexes, human NCO recombination maps for live offspring, along with complete recombination maps incorporating both COs and NCOs and maps of DSB resolution. These maps are an important tool for exploring the meiotic process and represent a major stepping-stone towards building a better understanding of the distribution of NCOs, DSB resolution, and the interplay between recombination and mutation, the two key processes underlying the generation of human genetic diversity. A better understanding of the recombination process may enable us to recognize the conditions under which this process fails and results in aneuploidies and pregnancy loss.
The numbers of NCOs and COs were positively correlated in both paternal and maternal meiosis; thus, the age increase observed in maternal COs1,55,56 is not due to DSBs being preferentially resolved as COs as the mother ages, but to an overall increase in meiotic DSBs in oocytes that result in successful pregnancies. Although the human maps for NCOs and COs are highly correlated, there are, as in other species6,17,57,58,59, genomic regions in which COs are clearly avoided—not because of a depletion of DSBs, but owing to DSB resolution. COs are likely to be suppressed in transcribed regions because of possible disruptive effects on the coding integrity of genes41, and in pericentromeric regions where their presence has been associated with meiotic segregation errors and aneuploidy49,60.
Chromosomal abnormalities may constitute a major cause of infertility and pregnancy loss as they are found in more than 50% of miscarriages and only 0.1% of live births61. Chromosomal abnormalities are mostly maternally transmitted, with abnormal placement of COs probably having a key role. We recently discovered62 that a missense variant in SYCE2 is associated with both pregnancy loss and several meiosis-related phenotypes, including distance of COs from telomeres1. SYCE2 is a key protein involved in the assembly of the synaptonemal complex backbone, the protein lattice that affects pairing of homologous chromosomes during meiosis. Proper segregation of chromosomes during meiosis depends on a tightly regulated placement of COs.
Paternal DSBs seem to be much more tightly regulated, as evidenced by the much higher fraction of oNCOs occurring within DMC1 (refs. 10,11) and CO1 hotspots. The difference becomes more prominent with maternal age, consistent with nearly all maternal-age-related increases in NCOs occurring outside DMC1 and CO hotspots. We postulate that different processes underlie age-related and other NCOs; however, our data do not reveal these processes.
DNMs are enriched near NCOs, with sex differences in both the range of impact on NCO-induced DNMs and the associated spectrum of DNMs. Paternal DNMs are primarily over-represented within 3 kb from an NCO, consistent with almost all paternal NCOs affecting a limited range around DSBs. By contrast, we observed elevation of the maternal DNM rate up to 100 kb. The total mutational contribution of DSBs is about 1.8% and 11.3% in paternal and maternal meiosis, respectively. For mothers, the contribution increases with age and is larger in CGER, for which our results indicate that the age-related increase in DNMs may be largely driven by NCOs.
The generation of new sequence diversity can be seen as an unwitting battle between the sexes. Mothers contribute mostly through COs, whereas fathers do so through DNMs. Fathers also contribute more through NCOs than mothers, in that they yield more of them, whereas NCOs from mothers are longer and therefore contain more MPPs. We show that recombination contributes a substantial fraction of the DNMs. The rate of DNMs is increased in the areas flanking DSBs, no matter whether they are resolved through CO or NCO or come from the mother or the father.
Methods
Data
To call gene conversions, we used whole-genome sequence data for parents and children in 2,132 Icelandic families with two or more children, comprising a total of 10,840 meioses. The dataset was made up of Icelandic samples collected as part of disease-association efforts at deCODE genetics and consisted of data for 173,025 SNP chip-typed individuals, of which a subset of 63,118 had been whole-genome sequenced63,64 (Supplementary Note 1). All participants were Icelanders who signed an informed consent form and donated biological samples for genotyping as part of various research projects at deCODE genetics approved by the National Bioethics Committee in Iceland. Data were processed in agreement with the approvals issued by the National Bioethics Committee, conditions set by the Data Protection Authority (ref. PV_2017060950ÞS) on procedures to ensure security in the processing of personal data for scientific research within the health sector conducted by deCODE genetics, and the Act on Scientific Research in the Health Sector no. 44/2014.
NCO calls
Gene conversions were detected in offspring by phasing the genotypes of both children and parents on a curated set of SNPs and indel variants. Phasing could be done on informative variants (MPPs), that is, those that were heterozygous in one parent and homozygous for the other. The phasing was trivial for the offspring as one of the parents was homozygous. The parents were then phased against the set of children. This phasing allowed us to assign a grandparental origin to haplotypes in the offspring and thus detect where the grandparental origin of haplotypes changed, indicating either COs or NCOs. Haplotypes that spanned less than 100 kb between such changes were regarded as gene conversion candidates and could be grouped into an oNCO if they were consecutive and did not form part of a CO (Supplementary Note 4).
Quality checks
The phasing of the parent at variants for which a candidate gene conversion transmission had been detected in offspring was verified with two approaches. Phasing was confirmed if variants of alternative grandparental origin in the offspring were close enough that they could be observed together on a sequence read for the parent. If the variants were further apart, we used the Icelandic genealogical database to find one or more relatives that shared, identical by descent with the parent, large haplotypes containing the variants to be checked. If those relatives happened to be also homozygous at those variants, we could use the relationship of the parent and the relative to establish whether the variant was paternally or maternally inherited (Supplementary Note 2.1).
We also benchmarked our methodology by comparison with earlier methods24 that used three-generation family structures with three or more siblings (Supplementary Note 3.4). All summary statistics were comparable (Supplementary Tables 13 and 14), with no unexplained discrepancies (Supplementary Table 15). Finally, to check whether family size affected our results, we verified that all summary statistics were comparable between families with only two children and larger families (Supplementary Table 16).
Length distributions, NCOs and DSBs
The length distributions of the NCOs (observed or not) were modelled as mixtures of negative binomial distributions using the NCOurd approach23 (Supplementary Note 4). NCOurd models the mismatch repair of single-stranded DNA at the site of NCOs and requires the complete set of oNCO along with the set of MPPs for each proband. The set of MPPs was used to compute a tract function for each oNCO and a detection probability function, which depended on the length. The mixture components of the resulting length distribution were separated into two groups: short and extended, having mean length under and over 1 kb, respectively.
The NCOurd results and the detection probability function were used to derive the expected fraction of NCOs detected as oNCOs. Using this detection fraction and the total number of oNCOs, we estimated the average number of NCOs (NNCO) per offspring; then, the average number of DSBs (NDSB) per meiocyte was estimated from NNCO and the average number of COs (NCO) per offspring (Supplementary Note 4.4) as follows:
Statistics, confidence intervals and P values
Unless otherwise specified, all statistics pertain to the data for the full cohort (possibly in groups) and the length distributions that were computed with the full cohort. Reported averages were computed using the full cohort, with confidence intervals computed by bootstrapping from the set of parents (Supplementary Note 11). Thus, quantities were computed per parent on the basis of the data for the children; then, statistics were computed for 1,000 bootstrap samples from the set of parents. The lower and upper limits of the confidence intervals correspond to the 2.5 and 97.5 percentile of the bootstrap statistics. The same bootstrap approach was used for computing matched pairs for estimating P values for comparison of two datasets (Supplementary Note 11).
An exception to this approach was used for the estimates of confidence intervals for length distribution parameters, and for the numbers of NCOs and NCO-derived quantities. For these, we computed 1,000 different length distributions by sampling data from the set of parents as described above. Reported values correspond to a computation done with the full cohort, and the confidence intervals were computed on the basis of the bootstrap estimates as outlined above.
All computed P values were two-sided.
NCO maps
NCO maps were computed on a grid of overlapping 3-Mb windows at intervals of 1 Mb (Supplementary Note 5). Self-consistent distributions for the number of paternal and maternal NCOs per window were derived on the basis of the number of oNCOs falling within the window and the expected NCO detection fraction in the window. The expected number of NCOs per window could then be obtained from those distributions.
DSB maps were created from the NCO and CO maps, with the DSB map value (nDSB) given in terms of the NCO map value (nNCO) and the CO map value (nCO) as:
We explored DSB resolution with a normalized NCO/CO difference map, ΔDSB, given by:
Here, NNCO and NCO indicate the average numbers of NCOs and COs per offspring, respectively. We note that ΔDSB takes the values 1 and −1 if all DSBs within a given map window are resolved as NCOs and COs, respectively, and 0 if the resolution conforms to the genome-wide average.
Analysis of telomere and centromere distances
Distances were measured from the centre of each map window. Distances to telomeres were measured to the ends of the chromosomes as given by GRCh38 (ref. 65). Distances to centromeres were measured to the edge of the centromere66,67. If the window centre was within the centromere, the distance was regarded as zero. Distances were binned in bins of size 1 Mb, and the x coordinate of each point in the graph was the centre of the corresponding bin.
DNMs
We explored the mutagenicity of NCOs by analysing the enrichment of DNMs near oNCOs (Supplementary Note 10); that is, we compared the observed numbers of DNMs in bins at different distances from oNCOs with the number of DNMs expected on the basis of the normative DNM rate. The normative DNM rate was computed per proband on the basis of the age of the parent at the birth of the proband. A Bayesian approach using a beta distribution prior was used to estimate the paternal–maternal split of unphased DNMs. We analysed whether the rate enrichment might be due to sequence context by computing the enrichment after permuting the DNMs among probands. No enrichment was found after permutation (P > 0.2), indicating that the observed enrichment was due to the mutagenicity of NCOs. We performed the enrichment analysis separately for each component of the NCO length distributions, enabling us to compute the final enrichment of DNMs near NCOs, weighted appropriately for NCOs rather than oNCOs. Finally, we estimated the contribution of NCOs to the overall number of DNMs by multiplying the total number of NCOs and the enrichment of the DNMs rate near NCOs.
DNM spectra were compared with a two-sided χ2 test. The χ2 statistic and P value were computed for the difference between the oNCO-proximal spectrum and the genome-wide spectrum for the probands in the study. The null distribution for the χ2 statistic was simulated by sampling one million times from the complete set of DNMs for the probands in the study. We sampled the same number of DNMs as in the oNCO-proximal spectrum, using data from the same number of DNM-transmitting parents as in that spectrum. For fathers, we found 74,950 events with χ2 statistic larger than that computed for the oNCO-proximal spectrum, giving a P value of 0.075. For mothers, no events were found with larger χ2 statistic than that for the oNCO-proximal spectrum. For the maternal distribution, we computed an inflation factor as the number of degrees of freedom (7) divided by the average of the χ2 statistic across the million simulations (6.96). As this factor (7/6.96 = 1.006) was greater than 1.0, we quote the unadjusted P value for the difference between the spectra.
Odds ratios (with P values and confidence intervals) for the strand asymmetry of oNCO-proximal DNMs were calculated using the fisher.test function of R68. For each class, we omitted DNMs transmitted by parents to two or more distinct children in the class. For the reported classes, this only affected the maternal C>T class, for which two DNMs were omitted from the calculation, with minimal effect on the P value and odds ratio. To determine whether the fraction of oNCO-proximal DNMs within a mutation class differed significantly from the genome-wide fraction shown in Fig. 1, we used bootstrapping (Supplementary Note 11) to compute an odds ratio and two-sided test to determine whether the bootstrap values differed significantly from 1.
Age effects
We performed linear regressions to analyse how parental age at birth of offspring affected various recombination statistics. For statistics that pertained to oNCOs and COs, for which we had an observed statistic per proband, we computed the linear regression directly on those data. For estimated statistics, such as the number of NCOs and DSBs, we split the cohort into groups on the basis of parental age and computed the average statistic for each group. Confidence intervals were estimated for each group using bootstrapping as described above. The linear regression was then performed on the average, weighted with the inverse of the estimated confidence interval.
Mixed model linear regression was used for the analysis of the DMC1 and maternal CO hotspot annotations of oNCOs. This was done with the lmerTest69 package in R68. Here, we computed the statistic for each proband and performed the regression on the complete set of data. Confidence intervals for linear regression results were computed on the basis of Student’s t-distribution within the packages used for computation69,70,71.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Recombination maps, proband information, gene conversions, oNCOs and DNMs are available at Zenodo (https://doi.org/10.5281/zenodo.14025564)72; GRCh38 at http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/; GIAB whole-genome sequencing samples at https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/; structural variant data of Beyter et al. at https://github.com/DecodeGenetics/LRS_SV_sets; and RefSeq at https://www.ncbi.nlm.nih.gov/refseq/. Eichler structural variant calls are shared on dbVar (https://www.ncbi.nlm.nih.gov/dbvar) under accession dbVar:nstd162. DMC1 hotspots are available at https://www.science.org/doi/suppl/10.1126/science.adh2531/suppl_file/science.adh2531_data_s1.zip. PRDM9 and H3K4me3 data of Altemose et al. at https://ftp.ncbi.nlm.nih.gov/geo/series/GSE99nnn/GSE99407/suppl/. Gnomad structural variants at http://gnomad-sg.org/downloads; and CO recombination data at https://www.science.org/doi/suppl/10.1126/science.aau1043/suppl_file/aau1043_datas1.gz and https://www.science.org/doi/suppl/10.1126/science.aau1043/suppl_file/aau1043_datas2.gz. The raw sequence data and the Icelandic genealogical database cannot be made publicly available because Icelandic law and the regulations of the Icelandic Data Protection Authority prohibit the release of individual-level and personally identifying data. Data access for raw data can be granted for scientific purposes only at the facilities of deCODE genetics in Iceland, subject to Icelandic law regarding data usage. Anyone wishing to gain access to the data should contact B.V.H. ([email protected]) or K.S. ([email protected]). Requests for access are generally considered monthly. Summary statistics for GWAS studies are available at https://www.decode.com/summarydata/.
Code availability
We used the following publicly available software in conjunction with the above-described algorithms: GraphTyper (v.2.7.1), https://github.com/DecodeGenetics/graphtyper; NCOurd, https://github.com/DecodeGenetics/NCOurd; R (v.4.2.2 with lm v.4.2.2, xoi v.0.67-1), https://www.r-project.org/; Python (v.3.8.1 with numpy v.1.24.2, pandas v.1.4.0, scipy v.1.10.1, statsmodels v.0.13.2), https://www.python.org/downloads/.
References
Halldorsson, B. V. et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, eaau1043 (2019).
Kong, A. et al. A high-resolution recombination map of the human genome. Nat. Genet. 31, 241–247 (2002).
Bhérer, C., Campbell, C. L. & Auton, A. Refined genetic maps reveal sexual dimorphism in human meiotic recombination at multiple scales. Nat. Commun. 8, 14994 (2017).
Broman, K. W., Murray, J. C., Sheffield, V. C., White, R. L. & Weber, J. L. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 63, 861 (1998).
Frazer, K. A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
Haber, J. Genome Stability (Garland Science, 2013).
Zickler, D. & Kleckner, N. Recombination, pairing, and synapsis of homologs during meiosis. Cold Spring Harb. Perspect. Biol. 7, 1–28 (2015).
Bergman, J. & Schierup, M. H. Evolutionary dynamics of pseudoautosomal region 1 in humans and great apes. Genome Biol. 23, 215 (2022).
Jónsson, H. et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 (2017).
Pratto, F. et al. Recombination initiation maps of individual human genomes. Science 346, 1256442 (2014).
Hinch, R., Donnelly, P. & Hinch, A. G. Meiotic DNA breaks drive multifaceted mutagenesis in the human germ line. Science 382, eadh2531 (2023).
Sun, H., Treco, D., Schultes, N. P. & Szostak, J. W. Double-strand breaks at an initiation site for meiotic gene conversion. Nature 338, 87–90 (1989).
Baudat, F. et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 (2010).
Robert, T. et al. The TopoVIB-like protein family is required for meiotic DNA double-strand break formation. Science 351, 943–949 (2016).
Keeney, S., Giroux, C. N. & Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384 (1997).
Szostak, J. W., Orr-Weaver, T. L., Rothstein, R. J. & Stahl, F. W. The double-strand-break repair model for recombination. Cell 33, 25–35 (1983).
Zickler, D. & Kleckner, N. Meiosis: dances between homologs. Annu. Rev. Genet. 57, 1–63 (2023).
Chen, J.-M., Cooper, D. N., Chuzhanova, N., Férec, C. & Patrinos, G. P. Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet. 8, 762–775 (2007).
Handel, M. A. & Schimenti, J. C. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 11, 124–136 (2010).
Gray, S. & Cohen, P. E. Control of meiotic crossovers: from double-strand break formation to designation. Annu. Rev. Genet. 50, 175–210 (2016).
Collins, J. K. & Jones, K. T. DNA damage responses in mammalian oocytes. Reproduction 152, R15–R22 (2016).
Gudbjartsson, D. F. et al. Sequence variants from whole genome sequencing a large group of Icelanders. Sci. Data 2, 150011 (2015).
Hardarson, M. T., Palsson, G. & Halldorsson, B. V. NCOurd: modelling length distributions of NCO events and gene conversion tracts. Bioinformatics 39, btad485 (2023).
Halldorsson, B. V. et al. The rate of meiotic gene conversion varies by sex and age. Nat. Genet. 48, 1377–1384 (2016).
Williams, A. L. et al. Non-crossover gene conversions show strong GC bias and unexpected clustering in humans. eLife 4, e04637 (2015).
Browning, S. R. & Browning, B. L. Biobank-scale inference of multi-individual identity by descent and gene conversion. Am. J. Hum. Genet. 111, 691–700 (2024).
Tiemann-Boege, I., Schwarz, T., Striedner, Y. & Heissl, A. The consequences of sequence erosion in the evolution of recombination hotspots. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160462 (2017).
Kostka, D., Hubisz, M. J., Siepel, A. & Pollard, K. S. The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol. 29, 1047–1057 (2012).
Jeffreys, A. J. & May, C. A. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat. Genet. 36, 151–156 (2004).
Wall, J. D., Robinson, J. A. & Cox, L. A. High-resolution estimates of crossover and noncrossover recombination from a captive baboon colony. Genome Biol. Evol. 14, evac040 (2022).
Versoza, C. J. et al. Novel insights into the landscape of crossover and noncrossover events in rhesus macaques (Macaca mulatta). Genome Biol. Evol. 16, evad223 (2024).
Cole, F. et al. Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat. Genet. 46, 1072–1080 (2014).
Mimitou, E. P., Yamada, S. & Keeney, S. A global view of meiotic double-strand break end resection. Science 355, 40–45 (2017).
Paiano, J. et al. ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis. Nat. Commun. 11, 1–15 (2020).
Oliver-Bonet, M., Campillo, M., Turek, P. J., Ko, E. & Martin, R. H. Analysis of replication protein A (RPA) in human spermatogenesis. Mol. Hum. Reprod. 13, 837–844 (2007).
Lenzi, M. L. et al. Extreme heterogeneity in the molecular events leading to the establishment of chiasmata during meiosis i in human oocytes. Am. J. Hum. Genet. 76, 112–127 (2005).
Wang, S. et al. Per-nucleus crossover covariation and implications for evolution. Cell 177, 326–338.e16 (2019).
Martini, E., Diaz, R. L., Hunter, N. & Keeney, S. Crossover homeostasis in yeast meiosis. Cell https://doi.org/10.1016/j.cell.2006.05.044 (2006).
Altemose, N. et al. A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis. eLife 6, e28383 (2017).
Boix, C. A., James, B. T., Park, Y. P., Meuleman, W. & Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021).
Kong, A. et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103 (2010).
Powers, N. R. et al. The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLoS Genet. 12, e1006146 (2016).
Pai, C. C. et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun. 5, 4091 (2014).
Baudat, F. & de Massy, B. Regulating double-stranded DNA break repair towards crossover or non-crossover during mammalian meiosis. Chromosom. Res. 15, 565–577 (2007).
Hinch, A. G. et al. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 363, eaau8861 (2019).
Centola, M. & Carbon, J. Cloning and characterization of centromeric DNA from Neurospora crassa. Mol. Cell. Biol. 14, 1510–1519 (1994).
Puechberty, J. et al. Genetic and physical analyses of the centromeric and pericentromeric regions of human chromosome 5: recombination across 5cen. Genomics 56, 274–287 (1999).
Mahtani, M. M. & Willard, H. F. Physical and genetic mapping of the human X chromosome centromere: repression of recombination. Genome Res. 8, 100–110 (1998).
Vincenten, N. et al. The kinetochore prevents centromere-proximal crossover recombination during meiosis. eLife 4, e10850 (2015).
Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993).
Chan, K. & Gordenin, D. A. Clusters of multiple mutations: incidence and molecular mechanisms. Annu. Rev. Genet. 49, 243–267 (2015).
Pratto, F. et al. Meiotic recombination mirrors patterns of germline replication in mice and humans. Cell https://doi.org/10.1016/J.CELL.2021.06.025 (2021).
Neri, F. et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77 (2017).
Goldmann, J. M. et al. Germline de novo mutation clusters arise during oocyte aging in genomic regions with high double-strand-break incidence. Nat. Genet. 50, 487–492 (2018).
Kong, A. et al. Recombination rate and reproductive success in humans. Nat. Genet. 36, 1203–1206 (2004).
Martin, H. C. et al. Multicohort analysis of the maternal age effect on recombination. Nat. Commun. 6, 7846 (2015).
Li, R. et al. A high-resolution map of non-crossover events reveals impacts of genetic diversity on mammalian meiotic recombination. Nat. Commun. 10, 3900 (2019).
de Boer, E., Jasin, M. & Keeney, S. Local and sex-specific biases in crossover vs. noncrossover outcomes at meiotic recombination hot spots in mice. Genes Dev. 29, 1721–1733 (2015).
de Boer, E., Stam, P., Dietrich, A. J. J., Pastink, A. & Heyting, C. Two levels of interference in mouse meiotic recombination. Proc. Natl Acad. Sci. USA 103, 9607–9612 (2006).
Wang, H. & Xu, X. Microhomology-mediated end joining: new players join the team. Cell Biosci. 7, 6 (2017).
Wartosch, L. et al. Origins and mechanisms leading to aneuploidy in human eggs. Prenat. Diagn. 41, 620–630 (2021).
Steinthorsdottir, V. et al. Variant in the synaptonemal complex protein SYCE2 associates with pregnancy loss through effect on recombination. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-023-01209-y (2024).
Kong, A. et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 40, 1068–1075 (2008).
Kong, A. et al. Parental origin of sequence variants associated with complex diseases. Nature 462, 868–874 (2009).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Cheung, V. G. et al. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature 409, 953–958 (2001).
Furey, T. S. & Haussler, D. Integration of the cytogenetic map with the draft human genome sequence. Hum. Mol. Genet. 12, 1037–1044 (2003).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2017).
Kuznetsova, A., Brockhoff, P. B. & Christensen, R. H. B. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017).
Seabold, S. & Perktold, J. Statsmodels: econometric and statistical modeling with Python. In Proc. 9th Python in Science Conference (eds van der Walt, S. & Millman, J.) 92–96 (SciPy, 2010).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Pálsson, G. DecodeGenetics/PalssonEtAl_Nature_2024: initial release of data. Zenodo https://doi.org/10.5281/zenodo.14025564 (2024).
Acknowledgements
We thank the study participants.
Author information
Authors and Affiliations
Contributions
The paper was written by G.P. and B.V.H. with input from M.T.H., H.J., V.S., O.A.S., H.P.E., U.T., P.S., A.H., D.F.G. and K.S. Gene conversions and NCOs were analysed by G.P. and M.T.H., and DNMs by H.J. Annotation analysis was by O.A.S. and S.A.G. Genotyping was performed by H.P.E.; imputation was performed by P.I.O. and A.G, supervised by G.M.; and statistical analysis was by G.P., M.T.H. and B.V.H., supervised by B.V.H. and D.F.G. The study was supervised by B.V.H. and K.S. All authors agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
All authors are employees of deCODE genetics/Amgen.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Co-conversion probability and oNCO extent.
a| Proportion of gene converted markers as function of distance from the first marker of oNCOs. b| Proportion of markers that are within the oNCO (gene converted or not) as a function of distance from the first marker of oNCOs. The fraction of markers within oNCOs for a given distance provides a lower bound on fraction of oNCOs shorter than that distance.
Extended Data Fig. 2 Variation of recombination maps with distance to nearest telomere.
The figure shows the average map values vs. distance to the nearest telomere, computed on a grid of 3 Mb overlapping windows and normalized individually with respect to their genome-wide mean. The x-coordinates for the telomere distance are shifted slightly in opposite directions for paternal/maternal meiosis so that error bars don’t overlap for nearby points. The error bars indicate 95% confidence intervals and are computed by bootstrap sampling 1000 times from the set of windows within each distance bin.
Extended Data Fig. 3 Variation of recombination maps with GC content.
The GC content is computed on the same overlapping 3 Mb windows as the maps, split into deciles and the average map values computed for the windows that fall into each decile. All maps except ΔDSB are normalized individually with respect to their genome-wide mean. The x-coordinate in the figures shows the median GC content in each decile, shifted slightly in opposite directions for paternal/maternal meioses so that errors bars don’t overlap for nearby points. The error bars indicate 95% confidence intervals and are computed by bootstrap sampling the map values 1000 times from the set of windows within each decile.
Extended Data Fig. 4 Variation of recombination maps with replication timing.
The replication timing is computed on the same overlapping 3 Mb windows as the maps, split into deciles and the average map values computed for the windows that fall into each decile. All maps except ΔDSB are normalized individually with respect to their genome-wide mean. The x-coordinate in the figures corresponds to the median replication time in each decile, shifted slightly in opposite directions for paternal/maternal meioses so that errors bars don’t overlap for nearby points. Early replication time corresponds to replication timing of 1.0, Mid corresponds to 0.0, and Late corresponds to −1.0. The error bars indicate 95% confidence intervals and are computed by bootstrap sampling the map values 1000 times from the set of windows within each decile.
Extended Data Fig. 5 Distribution of NCO-proximal DNMs.
The count of phased SNP/Indel DNMs near oNCOs vs. distance from the center of the oNCO.
Extended Data Fig. 6 Comparison of mutation spectra for phased DNMs.
DNMs are considered proximal to oNCOs and COs if they are within 3 kb and 100 kb for paternally and maternally phased DNMs, respectively. The length of bars indicates the mutation class fraction, computed for 5400 probands. Error bars represent 95% confidence intervals, computed using 1000 bootstrap samples.
Extended Data Fig. 7 Strand asymmetry spectra for SNP DNMs.
The count of phased SNP DNMs within the regions of enriched DNM rate around oNCOs, i.e. within 3 kb for paternal DNMs and within 100 kb for maternal DNMs. Strand asymmetry of DNM variants and their complement is observed in four mutation classes: maternal C > A (Fisher’s test p-value: 0.039), maternal C > G (Fisher’s test p-value: 2.3·10−6), and maternal C > T (Fisher’s test p-value: 7.0·10−5), and paternal CpG > TpG (Fisher’s test p-value: 4.7·10−3).
Supplementary information
Supplementary Information
Supplementary Notes 1–11, Figs 1–7, Tables 1–26 and references.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Palsson, G., Hardarson, M.T., Jonsson, H. et al. Complete human recombination maps. Nature 639, 700–707 (2025). https://doi.org/10.1038/s41586-024-08450-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-08450-5
This article is cited by
-
Rate of de novo mutations in the three-spined stickleback
Heredity (2025)
-
Sequence diversity lost in early pregnancy
Nature (2025)
