
Abstract
Several neurodegenerative disorders (NDDs), notably amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are characterized by pathological cytoplasmic aggregation of TAR DNA-binding protein 43 (TDP-43) in neurons and glia. Primarily localized in the nucleus under physiological conditions, TDP-43 is a critical regulator of RNA processing and metabolism. Therefore, RNA changes induced by TDP-43 depletion or mutation could play an important role in the pathogenesis of ALS and other TDP-43 related NDDs.To investigate these effects in NSC34 motor neuron-like cells, a commonly used cellular model of ALS, we used RNA interference to knock down TDP-43 and overexpressed the ALS-associated TDP-43 M337V mutation. RNA from both these experiments was enriched for small and large transcripts and subsequently analyzed via next-generation sequencing.The resulting transcriptomics datasets offer a valuable resource for studying the impact of TDP-43 depletion and mutant over-expression in motor neurons. These data enable comprehensive differential expression analyses and functional enrichment studies, identifying cellular pathways affected by TDP-43 depletion or mutation. Additionally, the inclusion of non-coding RNAs facilitates the construction of gene regulatory networks, providing insights into the interplay between coding and non-coding RNAs in gene expression regulation under TDP-43 loss-of-function or pathogenic mutation conditions.
Similar content being viewed by others


Background & Summary
TAR DNA-binding protein 43 (TDP-43) is a ribonucleoprotein implicated in several neurodegenerative disorders (NDDs)1. Primarily localized in the nucleus under physiological conditions, TDP-43 is a critical regulator of RNA transcription, splicing, stability, and metabolism2,3,4,5. However, in several NDDs, TDP-43 undergoes a pathological shift in its localization to the cytoplasm6,7,8,9. Cytoplasmic aggregates of TDP-43, which can be phosphorylated and ubiquitinated, are observed in the brain and spinal cord in various NDDs, such as amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), Alzheimer’s disease (AD) and Parkinson disease8,10. Notably, roughly 97% of ALS and 45% of FTLD patients show neuronal and glial inclusions containing TDP-43 with significant nuclear clearance of the protein5.
While emerging evidence supports a role of TDP-43 in RNA processes, understanding the mechanisms by which it contributes to neurodegeneration is complex. The TARDBP gene, responsible for encoding the TDP-43 protein, harbours over 50 pathogenic mutations associated with both sporadic and familial forms of ALS and FTLD4,11,12. These mutations predominantly cluster in the amyloidogenic C-terminal glycine-rich region of the protein, rendering TDP-43 prone to forming amyloid-like fibrils thus enhancing its propensity for aggregation5. One such mutation commonly found in ALS patients is the TDP-43M337V mutation where Methionine 337 is substituted by Valine13. This mutation promotes TDP-43 cytoplasmic mislocalization and disrupts TDP-43 regulated RNA processes ultimately leading to widespread transcriptomic dysregulation5,12,14. Notably, several of these changes overlap with those observed in TDP-43 knockdown models, suggesting a shared mechanism of RNA dysregulation5,11.
While the evidence indicates broad transcriptomic changes in various neuronal models after TDP-43 depletion and mutant expression, a parallel comparison of TDP-43 depletion and mutant conditions in the same cellular model is unprecedented. Furthermore, most transcriptomic studies in TDP-43 models have traditionally focused on either the coding or non-coding regions of the transcriptome, with limited consideration of how changes in one affect the other. Here, employing a library preparation method that captures both coding and non-coding fractions of RNA, we have generated comprehensive transcriptomic datasets following TDP-43 knockdown and mutant TDP-43M337V expression, as well as their respective controls, in NSC34 motor neuron-like cells. These datasets are a valuable resource for elucidating how TDP-43 loss of function or mutation can perturb the coding and non-coding transcriptome in a commonly used cellular model of motor neurons. Moreover, it facilitates the comprehension of how alterations in the non-coding transcriptome impact the coding transcriptome, and vice versa.
Methods
Cell culture
NSC34 motor neuron-like cells (Tebubio: CLU140-A) were cultured in DMEM culture medium (Sigma-Aldrich: D5796) supplemented with 10% Serum (Gibco: 10500064) and 1% Penicillin-Streptomycin (Gibco: 15140122) at 37 °C in a humidified incubator with 5% CO2. Twenty-four hours before transfection, cells were seeded to a confluency of 60% in a 6-well plate.
TDP-43 Knockdown in NSC34 motor neurons
TDP-43 expression was depleted in NSC34 motor neurons using RNA interference (Fig. 1a). NSC34 motor neurons were transfected with two TDP-43 siRNAs: Mm_Tardp 1 siRNA (AACGATGAACCCATTGAAATA, Qiagen: SI01441503|S2) and Mm_Tardp 2 siRNA (CAGTAACATGGTAACATTAAA, Qiagen: SI01441510|S2) as the TDP-43 knockdown group (TDP-43-KD, n = 8). The non-silencing (NS) siRNA (Qiagen: 1027281) was used as a control (n = 8). On the day of transfection, Lipofectamine RNAiMAX reagent(Invitrogen™:13778075) and siRNAs were individually diluted in Opti-MEM® Medium. These diluted components were then combined at a 1:1 ratio and incubated for 10 minutes to form the siRNA-lipid complex. The resulting complex was added to fresh media to achieve a final siRNA concentration of 20 nM. Subsequently, cells were incubated under standard conditions for 6 hours, after which their medium was replaced with fresh growth medium. Cells were then further incubated under standard conditions for up to 24 hours before proceeding to RNA extraction.

RNA-Seq workflow and analysis pipeline. (a) Steps for RNA-seq library preparation, including RNA extraction, quality checks, and library construction for both experimental settings: TDP-43 Knockdown (KD) with its respective control (NS), and TDP-43 mutant expression (MVT) with its respective control (WTT). (b) Bioinformatics pipeline for processing raw FASTQ data, from quality trimming to gene expression quantification, including the software tools used. (c) Workflow for read normalization, sample quality control, and gene biotype annotation as reported in this manuscript.
TDP-43 wildtype and mutant synthesis and plasmid expression in NSC34 motor neurons
The open reading frame of wild-type TDP-43 and mutant TDP-43M337V were synthesized and cloned into the Express Cloning Vector pcDNA3.1 + C-eGFP generating the Cloning vectors: TDP-43_WTT_pcDNA3.1( + )-C-eGFP plasmid (Genescript: SC1691) and TDP-43_MVT_pcDNA3.1( + )-C-eGFP (Genescript: SC1693), from here on referred to as WTT (Wild type TDP-43), and MVT (mutant TDP-43), respectively. Following synthesis and verification, WTT and MVT plasmids were transformed into E. coli and grown overnight. Three single colonies for each plasmid were selected, cultured in LB media with ampicillin, and subjected to mini-prep extraction using the Zyppy™ Plasmid Miniprep Kit (Zymo Research: D4036). The extracted plasmids were assessed for DNA concentration and purity using a Nanodrop Spectrophotometer and then analysed using long-read Sanger sequencing. Sequencing results were aligned with the original sequences using SnapGene for verification. Verified colonies were used to generate maxi-preps, ensuring high concentrations of the plasmid vector with the desired DNA sequences. For transfection, NSC34 cells were seeded to 60% confluency in 6-well plates and then transfected with WTT (control, n = 8) and MVT (mutant TDP-43, n = 8) using Lipofectamine™ 3000 Transfection Reagent (Invitrogen: L3000001) diluted in Opti-MEM. Plasmid DNA plus P300 reagents were also diluted in Opti-MEM. The diluted DNA and transfection reagent were mixed and added to fresh media. After incubation under normal conditions for up to 48 hours, transfection efficiency was assessed by observing green fluorescence before proceeding to RNA extraction.
RNA extraction and quality assessment
The culture media was removed and the cells were washed with ice-cold PBS. 1 ml of Trizol (Life Technologies: 15596018) was then added to each well, and plates were shaken for 5 minutes at room temperature on a rotary shaker at 300 RPM. The cell lysate was subsequently pipetted multiple times, transferred to a 1.5 ml centrifuge tube, and vigorously vortexed. After a 5-minute incubation at room temperature, 200 µl of chloroform was added to the lysate, vortexed, and incubated for 3 minutes at room temperature. Following centrifugation at 12,000 g for 15 minutes at 4 °C, 400 µl of the upper aqueous phase was transferred to a fresh 1.5 ml centrifuge tube, and an equal volume of chloroform was added. After another centrifugation at 12,000 g for 15 minutes at 4 °C, 400 µl of the upper aqueous phase was transferred to another fresh 1.5 ml centrifuge tube and 500 µl of isopropyl alcohol was added to the samples, mixed, and incubated at room temperature for 10 minutes. The samples were then centrifuged at 12,000 g for 10 minutes at 4 °C. After removal of the supernatant, the RNA pellet was washed twice with 1 ml 70% ethanol, followed by centrifugation at 7,500 g for 5 minutes. The pellet was then air-dried for 30 minutes, and the RNA resuspended in 30 µl DEPC-treated water. RNA quantification was performed using a Nanodrop Spectrophotometer. The integrity of total RNA was assessed using the Agilent 2100 Bioanalyzer with the RNA 6000 Nano Kit (Agilent Technologies, Ltd.). Libraries were exclusively prepared for samples with an RNA Integrity Number (RIN) greater than 9.0 (Fig. 2).

RNA quality assessment. RNA quality was evaluated across all experimental conditions, including non-silencing control (NS), TDP-43 knockdown (TDP43-KD), wild-type TDP-43 expression (WTT), and mutant TDP-43M337V expression (MVT). Electropherograms display characteristic ribosomal RNA peaks (18S and 28S), and the corresponding RNA Integrity Number (RIN) values are reported for each sample. All samples exhibit RIN values above 9, confirming high RNA integrity and suitability for RNA-seq library preparation.
Library preparation and RNA-sequencing
Library preparation was performed using the SEQuoia Complete Stranded RNA Library Prep Kit (Bio-Rad, cat. No. 17005726) according to the manufacturer’s protocol. Ribosomal depletion was achieved using the SEQuoia Ribodepletion Kit (Bio-Rad, cat. no. 17006487) as per the manufacturer’s instructions. Libraries were then constructed from 500 ng of total RNA. The procedure involved RNA fragmentation, end repair, addition of adenosines to the 3′ ends, and adapter addition with cDNA synthesis. Subsequently, cDNA fragments containing adapters were amplified by PCR. Library quality assessment was performed using the Agilent 2100 Bioanalyzer with the Agilent DNA High Sensitivity chip (Agilent Technologies, Ltd.), with a mean library size of 300 bp. Quantification of libraries was conducted using a Quantus fluorometer and QuantiFluor double-stranded DNA System (Promega, Madison, Wisconsin, USA). Sequencing was carried out as single-end 1 × 75 bp reads on the NovaSeq 6000 platform (Illumina, San Diego, USA) to generate approximately 50 million reads per library.
Quality control of sequencing reads
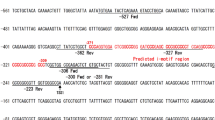
Raw data files - Binary Base Call (BCL) files generated by the Illumina sequencer (NovaSeq 6000) were converted using Bcl2Fastq tool (v2.20.0.422)15 to the text-based sequence file format that stores both raw sequence data and quality scores (FASTQ). The preprocessing of FASTQ files was performed using SeqSense NGS Data Analysis Toolkit from Bio-Rad (version 1.0)16 with the default parameters and with the option–skipUMI to skip the deduplication step. All the tools used in the implemented pipeline are presented in Fig. 1. The RNA-seq data were subjected to a series of quality control steps to ensure high-quality reads for downstream analysis. Initially, the raw FASTQ files were assessed using FASTQC (version 0.11.7)17 to evaluate the quality of the sequencing reads. Parameters such as per base sequence quality, GC content, and adapter content were inspected to identify potential issues (Fig. 3). Next, adapter sequences and low-quality bases were trimmed using Cutadapt (version 2.3)18 with Python 3.6.9. The following parameters were used: a minimum quality score of 0, a minimum read length of 15 bases, with an adapter sequence consisting of 10 repetitions of adenine to be trimmed, and the first nucleotide from the reads to be removed. This step removed residual adapter contamination and improved the overall quality of the reads (Fig. 3).

Overview of sequencing quality control. The left plots correspond to the KD vs. NS experiment, while the right plots correspond to the MVT vs. WTT experiment. (a) Per-base sequence quality scores demonstrate consistently high read-quality across the entire read length for all samples in both experiments, with most bases having Phred scores ≥ 30. (b) Per-sequence Phred quality scores show that the majority of reads exhibit high sequence quality, with Phred scores predominantly above 30, ensuring reliable base calls across all samples. (c) Sequence count distributions illustrate the estimated proportions of unique (blue) and duplicate (black) reads in each sample. The majority of reads are unique, confirming high sequencing depth and library complexity suitable for downstream differential expression analyses.
Alignment and quantification
The trimmed reads were then aligned to the mouse reference genome GRCm38/mm1019 using STAR (version 2.7.0 f)20. STAR was configured with default parameters. Post-alignment, SAM files were converted to BAM format, sorted, and indexed using Picard (version 2.20.0)21. The Picard tools were also used to mark duplicate reads to reduce bias in the downstream quantification steps. Bedtools (version 2.28.0)22 was used for the intersection of the reads that align within an annotated BED file containing known small and long RNAs (Fig. 1b).
Gene expression quantification on the small and long RNA BAM files with appropriate RNA annotation set was performed using the featureCounts function from the Subread package (version 1.6.4)23, exporting raw counts, and normalised expression in RPKMs and TPMs. The aligned reads were assigned to mouse genes annotated in the GRCm38 genome assembly. The resulting expression matrix was used for subsequent bioinformatics analyses (Fig. 1c). The MultiQC tool (version 1.14)24 was used in the SeqSense NGS Data Analysis Toolkit outputs to produce a merged report with the sequence’s quality.
RNA expression profiling
A quality control analysis of gene expression data was conducted, and the expression profiles for each experimental group were briefly examined using a customized pipeline of R scripts (R version 4.4.0)25. For this analysis, several R packages were used for data tidying, wrangling, and visualization, including tidyverse (version 2.0.0)26, DESeq2 (version 1.44.0)27, pheatmap (version 1.0.12)28, and patchwork (version 1.2.0)29.
To evaluate the overall similarity between samples, sample-level quality control was performed using two unsupervised clustering methods: Principal Component Analysis (PCA) and hierarchical clustering (Fig. 4). Both analyses were based on log2-transformed normalized counts to enhance distance metrics and clustering clarity for visualization. To mitigate bias from low-count genes, a regularized log transformation (rlog) was applied instead of a standard log2 transformation, as implemented by the rlogTransformation() function in the DESeq2 package (additional details on this approach are available in the DESeq2 vignette).

Sample variation and expression profile of TDP-43. (a) Principal Component Analysis (PCA) plot illustrating the clustering of samples based on gene expression profiles. Samples group according to their experimental conditions (NS, KD, WTT, and MVT), indicating consistent variation and clear segregation between groups, supporting the robustness of the dataset. (b) Heatmap of hierarchical clustering based on sample correlation coefficients. Strong intra-group similarity and distinct inter-group differences confirm the expected transcriptomic variation between conditions, validating the experimental design. (c) Bar plot of Tardbp (TDP-43) expression across experimental conditions. KD samples exhibit the lowest TDP-43 expression, confirming effective gene knockdown, while WTT and MVT samples show elevated expression, consistent with overexpression constructs. The NS control group exhibits intermediate endogenous expression levels, aligning with expected modulation based on genetic interventions.
Data Records
All sequencing data have been deposited in the ArrayExpress data repository with accession number E-MTAB-1373830. Metadata, including detailed experimental procedures, are also available at this repository. This dataset consists of 32 RNA sequencing raw data files in FASTQ format, corresponding to 8 replicates per experimental condition (NS, KD, WTT, and MVT). These raw reads are accessible via the European Nucleotide Archive (ENA) under accession number PRJEB7219931. Processed data (in plain text tabular format) containing counts and TPM normalised expression per gene feature (mouse genome annotation GRCm38) are deposited in Figshare32.
Technical Validation
Validation of experimental design strategy
The experimental design was assessed for any sources of variation and potential outliers by testing the sample variance via PCA (Fig. 4a). The first and second components accounted for 75% of the variance (67% in PC1 and 8% in PC2), clearly separating the four experimental groups into distinct clusters without overlap between replicates. This result supports the relevance of this dataset to test for differential expression between the experimental groups. Similarly, the hierarchical clustering of the sample pairwise correlation matrix (Fig. 4b) aligns with the experimental design, reinforcing the consistency across replicates.
Additionally, the expression levels of the Tardbp gene across the samples confirmed robust implementation of the experimental manipulations (Fig. 4c). As expected, the knockdown (KD) samples exhibit the lowest expression of the target gene. Conversely, the cells containing overexpression vectors (both the wild-type protein (WTT) and the mutant protein (MVT)) showed the highest expression values. The control group (NS), where the cells express endogenous gene levels, displays intermediate Tardbp expression values.
Validation of expression profiling
To validate the quantification and normalization steps, we assessed the read count distributions before and after TPM normalization, comparing the gene expression profiles across the four experimental groups (Fig. 5, top plot: before normalization; bottom plot: after normalization). The results clearly demonstrate that the samples share a consistent overall expression distribution post-normalization, confirming their suitability for meaningful future gene expression comparisons (e.g. differential expression analysis).

Impact of read count normalization. In both panels, the top plots represent raw expression data, while the bottom plots show data after TPM (transcripts per million) normalization. All values are log2-transformed for better visualization. (a) Boxplots of log2(counts) and log2(TPM) show gene expression distribution across samples before and after normalization. The consistent median expression levels across conditions indicate effective normalization, ensuring comparability across samples. (b) Density plots of log2(counts) and log2(TPM) illustrate the impact of normalization on expression distributions. The improved overlap across groups in the bottom panel demonstrates greater distribution consistency, reducing batch effects and making samples suitable for differential expression analysis.
Coding and non-coding gene expression
To validate the ribosomal depletion RNA-seq protocol and confirm the presence of both coding and non-coding genes in the dataset, we performed a biotype annotation count. The results, presented in Fig. 6 confirm the expected representation of both coding and non-coding genes. The bar plot highlights the distribution of the most frequent biotypes, classified into four groups: protein-coding genes, processed pseudogenes, and long non-coding RNAs (lncRNAs), with less abundant non-coding biotypes grouped under “other non-coding” RNA species. The inclusion of non-coding genes in this dataset provides an opportunity to explore both the transcriptomic landscape and the regulatory mechanisms of gene expression within the same dataset.

Biotype annotation counts. Distribution of coding and non-coding gene biotypes among the analyzed features. Protein-coding genes represent the majority (17,148 genes), followed by processed pseudogenes (2,869 genes), long non-coding RNAs (lncRNAs, 2,040 genes), and other non-coding RNAs (328 genes). In total, 22,385 genes were annotated by biotype out of 35,144 total genes present in the dataset, highlighting the inclusion of both coding and non-coding transcriptomic features.
Usage Notes
The gene expression data presented here provide a valuable resource for investigating the effects of TDP-43 depletion and mutant expression in NSC-34 cells, a widely used cellular model of ALS and related NDDs. This dataset supports differential expression analysis, enabling researchers to identify key cellular functions affected by the depletion, as well as overexpression of wild-type or mutant TDP-43. Importantly, the inclusion of non-coding RNAs expands the analytical potential by allowing the construction of comprehensive gene regulatory networks. This approach offers deeper insights into the interplay between coding and non-coding RNAs in regulating gene expression in motor neurons following TDP-43 loss-of-function or mutation.
Code availability
The software tools used to analyse this RNA-seq dataset were the following: FastQC (version 0.11.7)17, Cutadapt (version 2.3)18, STAR (version 2.7.0 f)20, Picard tools (version 2.20.0)21, BedTools (version 2.28.0)22, the Subread package (version 1.6.4)33, and MultiQC (version 1.14)24, as described in detail in the Methods section. Technical validation and data visualisation was performed in RStudio Server (version 2023.12.1 + 402), using R (version 4.4.0), and Bioconductor (version 3.19). R packages and further details can be found in the Methods section. The R code used for these analyses presented in the technical validation section is freely available in GitHub wrapped in an R package (https://github.com/patterninstitute/tdp43.nat.sci.data) in line with the guidelines and best practices for open and reproducible data analysis.
References
Liao, Y.-Z., Ma, J. & Dou, J.-Z. The Role of TDP-43 in Neurodegenerative Disease. Mol Neurobiol 59, 4223–4241 (2022).
Dash, B. P., Freischmidt, A., Weishaupt, J. H. & Hermann, A. An integrative miRNA-mRNA expression analysis identifies miRNA signatures associated with SOD1 and TARDBP patient-derived motor neurons. Human Molecular Genetics ddae072 (2024).
Wood, H. TDP43 blocks misprocessing of STMN2 RNA. Nat Rev Neurol 19, 326 (2023).
Christoforidou, E. et al. The ALS-associated TDP-43M337V mutation dysregulates microglia-derived extracellular microRNAs in a sex-specific manner. Disease Models & Mechanisms dmm-050638 (2024).
Tziortzouda, P., Van Den Bosch, L. & Hirth, F. Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat Rev Neurosci 22, 197–208 (2021).
Appleby‐Mallinder, C. et al. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathology and Applied Neurobiology 47, 61–72 (2021).
de Boer, E. M. J. et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry 92, 86–95 (2020).
Geser, F. et al. Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115, 133–145 (2008).
Huie, E. Z. et al. TDP-43 Pathology in the Setting of Intermediate and High Alzheimer’s Disease Neuropathologic Changes: A Preliminary Evaluation Across Ethnoracial Groups. Journal of Alzheimer’s Disease 1–11, https://doi.org/10.3233/JAD-220558 (2023).
TDP43 pathology in the brain, spinal cord, and dorsal root ganglia of a patient with FOSMN | Neurology. https://www.neurology.org/doi/10.1212/WNL.0000000000007008.
Konopka, A. et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Molecular Neurodegeneration 15 (2020).
Guerrero, E. N. et al. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Human Molecular Genetics 28, 2459 (2019).
Sreedharan, J. et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 319, 1668–72 (2008).
Jiang, L.-L. et al. Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci Rep 6, 23928 (2016).
bcl2fastq Conversion Software. https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html.
Jones, L. SeqSense Analysis Toolkit.
FastQC: A quality control tool for high throughput sequence data – ScienceOpen. https://www.scienceopen.com/book?vid=79473d05-c54d-43e6-b1ec-84c5e1991ce5.
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10 (2011).
Mus musculus genome assembly GRCm38. NCBI https://www.ncbi.nlm.nih.gov/data-hub/assembly/GCF_000001635.20/.
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Picard Tools - By Broad Institute. http://broadinstitute.github.io/picard/.
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (2024).
Wickham, H. et al. Welcome to the Tidyverse. JOSS 4, 1686 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Kolde, R. pheatmap: Pretty Heatmaps (2019).
Pedersen, T. L. patchwork: The Composer of Plots (2024).
Gbadamosi, I. et al. ArrayExpress. https://identifiers.org/arrayexpress:E-MTAB-13738 (2025).
ENA European Nucleotide Archive. https://identifiers.org/ena.embl:PRJEB72199 (2025).
Gbadamosi, I. et al. A Dataset of NSC34 Motor Neuron Coding and Non-coding Transcriptome Following TDP-43 Knockdown and Mutant TDP-43 M337V Expression. figshare. Dataset. https://doi.org/10.6084/m9.figshare.25898785.v1 (2024).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Research 41, e108–e108 (2013).
Acknowledgements
This project is supported by the EU Joint Programme–Neurodegenerative Disease Research (JPND) (TREMENDOS; UMO-2022/04/Y/NZ5/00122). RM and ID are supported by the Portuguese Fundação para a Ciência e Tecnologia (FCT) grant 2023.10973.CPCA. ID also receives additional support from the FCT through grant 2021.00238.CEECIND. Figure 1 was designed with https://app.biorender.com.
Author information
Authors and Affiliations
Contributions
I.G. designed and executed the experiments, performed the RNA extraction and quality checks, and led the drafting of the manuscript. S.B. performed bioinformatics data analysis and assisted in drafting the manuscript. B.G. performed the library preparations and sequencing. R.M. performed data quality control analysis, provided data analysis consulting, R Package development, and helped in drafting the manuscript. I.D. provided data analysis consulting, performed data analysis, and helped in drafting the manuscript. A.J. was responsible for securing the funding, the conceptualization and strategic oversight of the study, and reviewing the final draft of the manuscript. All authors have read and approved the final manuscript draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gbadamosi, I., Binias, S., Gielniewski, B. et al. Coding and non-coding RNA expression in NSC34 cells following TDP-43 depletion and mutant TDP-43 M337V expression. Sci Data 12, 1110 (2025). https://doi.org/10.1038/s41597-025-05409-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05409-7
