
Abstract
Kidney renal clear cell carcinoma (KIRC), a cancer characterized by substantial immune infiltration, exhibits limited sensitivity to conventional radiochemotherapy. Although immunotherapy has shown efficacy in some patients, its applicability is not universally effective. Studies have indicated that programmed cell death (PCD) can modulate the activity of immune cells and participate in the regulation of antitumor immune responses. However, systematic research on how various PCD patterns in KIRC affect the responsiveness to immunotherapy is lacking and requires in-depth investigation. We utilized a combination of 101 machine learning algorithms to analyze the TCGA-KIRC cohort and the GSE22541 KIRC patients, screening for cell death patterns closely associated with prognosis from 18 potential modes. Integrating multi-omics analysis, including immune cell infiltration, phenotyping, functional analysis, immune checkpoint exploration, and gene set enrichment analysis (GSEA), we explored the relationship between key cell death patterns and patients’ responses to immunotherapy. Finally, potential drug targets were identified through drug sensitivity screening and molecular docking techniques. Our sophisticated risk assessment model successfully identified two PCD patterns, Anoikis and lysosome-dependent cell death (LDCD), closely associated with the prognosis of KIRC patients, with the high-risk group exhibiting poor outcomes. Immune cell analysis revealed upregulated expression of T follicular helper (Tfh) cells in both PCD patterns. Analysis of immune checkpoints disclosed enhanced expression of human leukocyte antigen E (HLA-E) across both patterns. Frequent mutations in the TTN and MUC16 genes were observed in the Anoikis pattern, whereas in the LDCD pattern, although the high-risk group had a higher mutation rate, there was no significant difference in tumor mutational burden. GSEA analysis indicated significant enrichment of the primary immunodeficiency pathway in the Anoikis high-risk group and significant enrichment of the spliceosomal tri-snrnp complex assembly pathway in the LDCD high-risk group. Drug sensitivity analysis showed notable sensitivity to SB505124 in both PCD patterns. HMOX1 and PIK3CG were identified as common genes in the two key PCD patterns, and molecular docking analysis confirmed stable binding affinity between Carnosol and HMOX1, and between PROTAC and PIK3CG. Our study identifies Anoikis and LDCD as prognostic PCD patterns in KIRC, with key immune cells, genetic mutations, and drug sensitivity profiles. HMOX1 and PIK3CG are common genes with stable binding to Carnosol and PROTAC, respectively, while SB505124 shows significant sensitivity to both PCD modes, suggesting potential therapeutic targets.
Similar content being viewed by others
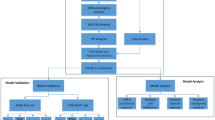


Introduction
Renal cancer is one of the most prevalent malignant tumors globally, with its incidence and mortality rates steadily increasing. It is projected that by the year 2040, the number of deaths attributable to this disease may soar to over 300,000 cases1,2. In terms of gender differences, the mortality rate has increased by 2.85% in males, while in females it has risen by 1.25%3. In China, approximately 50,088 new cases of renal cancer are diagnosed annually, with about 46,345 cases resulting in death4. Among all renal cancer cases, renal cell carcinoma accounts for up to 90%, with KIRC constituting 70%5. The prognosis of KIRC is generally grim, and the recurrence rate is as high as 30% for patients who have undergone surgical treatment6. Studies indicate that adjuvant therapy following surgery for localized renal cell carcinoma has always been a challenging research ___domain with limited success rates7. It is evident that the treatment of KIRC is a significant challenge in the global public health sector.
Malignant tumors of the urinary and reproductive systems, including prostate cancer8 and bladder cancer9, have traditionally been managed primarily through radiotherapy and chemotherapy. However, KIRC stands out due to its resistance to conventional chemotherapeutic agents. KIRC is one of the highly immune-infiltrated cancers10. Within the tumor microenvironment (TME), immune cells play a pivotal role in tumor growth, invasion, migration, and modulation of anti-tumor immunity11. Under these circumstances, immunotherapy has become an essential component of its therapeutic strategy12. Nevertheless, the combined application of immunotherapy and immune checkpoint inhibitors has shown positive effects only in certain patient populations13,14. Current research suggests that the variability in patients’ responses to immunotherapy may be associated with the tumor’s immune evasion mechanisms, which prevent some patients from benefiting from immune checkpoint blockade therapy15,16,17. Therefore, a deeper investigation into the mechanisms of tumor immune evasion is crucial for the development of more effective treatment strategies.
PCD is an orderly process of cell self-destruction that is precisely regulated at the genetic level and carried out through a series of specific molecular mechanisms to perform internal cleansing tasks, eliminating damaged or unnecessary cells to maintain the health and function of tissues18. PCD encompasses a range of different pathways of cell death, including apoptosis, pyroptosis, ferroptosis, autophagy, necroptosis, cuproptosis, parthanatos, entotic cell death, netotic cell death, LDCD, alkaliptosis, oxeiptosis, NETosis, immunogenic cell death, Anoikis, paraptosis, methuosis, and entosis. Pyroptosis is a form of cell death process that triggers cell lysis and the release of inflammatory cytokines upon the activation of the inflammasome. Apoptosis is a form of programmed cell death regulated by caspases, characterized by significant changes in the nucleus and organelles, the blebbing of the plasma membrane, and the eventual fragmentation of the cell. Ferroptosis is an atypical form of programmed cell death closely related to iron-dependent lipid peroxidation. Autophagy is the process by which cells degrade their own damaged, aged, or superfluous biomolecules and organelles through lysosomes, a normal cellular self-renewal mechanism that releases free small molecules for cellular recycling. Necroptosis is a form of programmed cell death triggered by the receptor-interacting protein kinase signaling pathway in response to certain exogenous or endogenous stimuli, such as tumor necrosis factor. Cuproptosis is a cell death process directly induced by copper ions. Parthanatos is a form of programmed cell death caused by the overactivation of PARP-1, a DNA repair enzyme. Entotic cell death, also known as entosis, is the process where a living cell is engulfed by a conspecific cell and dies within the engulfing cell. Netotic cell death is a form of cell death driven by NETs, regulated by NADPH oxidase-mediated ROS production and histone citrullination. LDCD is a form of programmed cell death characterized by lysosomal dysfunction or rupture. Alkaliptosis is a specific form of cell death driven by intracellular alkalinization. Oxeiptosis is a form of atypical apoptosis-like cell death induced by reactive oxygen species that is caspase-independent. Immunogenic cell death is a unique form of cell death driven by stress that can activate the immune system to combat tumors or infections, often resulting from various cancer treatments such as radiation and chemotherapeutic drugs.Anoikis is a special form of programmed cell death induced when cells lose normal adhesion to the extracellular matrix and other cells. Paraptosis is a unique form of programmed cell death characterized by vacuolization of the cytoplasm, swelling of the mitochondria, and expansion of the endoplasmic reticulum. Methuosis is due to the dysregulation of large phagocytic vacuoles within the cell, leading to the formation of numerous vacuoles in the cytoplasm that coalesce and expand, ultimately causing cell death19,20,21,22.
PCD plays an extremely critical role in maintaining normal physiological activities and human health. It is noteworthy that PCD also plays an important role in the development of cancer. Studies have shown that PCD can regulate the enrichment of effector immune cells or regulatory immune cells, thereby participating in the regulation of anti-tumor immunity within the TME23. It can be seen that PCD plays a vital role in regulating the immune-suppressive TME and determining the clinical outcomes of cancer treatment methods. Current research indicates that there is a complex association between PCD and the response to and tolerance of immunotherapy in KIRC. For instance, in KIRC, a prognostic risk model related to ferroptosis reveals that patients in the high-risk group are more sensitive to immunotherapy24. Furthermore, a prognostic risk model constructed based on five genes associated with immunogenic cell death not only maps the tumor immune microenvironment of KIRC patients but also helps to promote the implementation of personalized treatment and provides potential new targets for immunotherapy25. These findings emphasize the importance of intervention strategies targeting PCD in KIRC immunotherapy.
Despite advances in understanding PCD pathways and immunotherapy in cancer, two critical gaps remain: (1) Most studies in KIRC rely on single-omics analyses or examine individual PCD mechanisms, overlooking the synergistic effects of multi-omics-driven PCD networks; (2) The potential of PCD patterns as biomarkers for immunotherapy stratification remains underexplored. To address these limitations, we propose a machine learning framework that integrates genomics, transcriptomics, and clinical data to decode pan-PCD patterns and their therapeutic implications in KIRC. This approach not only identifies novel immunotherapy targets but also provides a roadmap for personalized treatment strategies.
Methods
Data collection
We collected 18 modes of PCD and their key regulatory genes through literature search20. These genes include 580 genes related to apoptosis, 52 genes related to pyroptosis, 88 genes related to ferroptosis, 367 genes related to autophagy, 101 genes related to necroptosis, 19 genes related to cuproptosis, 9 genes related to parthanatos, 15 genes related to entotic cell death, 8 genes related to netotic cell death, 220 genes related to LDCD, 7 genes related to alkaliptosis, 5 genes related to oxidative stress-induced cell death, 24 genes related to NETosis, 34 genes related to immunogenic cell death, 338 genes related to anoikis, 66 genes related to paraptosis, 8 genes related to methuosis, and 23 genes related to phagoptosis, totaling 1964 genes, for analysis in this study. Additionally, TCGA-KIRC cohort data were downloaded from the TCGA database. RNA sequencing data and corresponding clinical pathological information of 614 KIRC samples were downloaded from the TCGA database (https://portal.gdc.cancer.gov/projects/TCGA-KIRC). Clinical pathological information and microarray expression analysis data of the KIRC cohort GSE22541 were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
Identification of differentially expressed genes (DEGs) in KIRC
RNA sequencing data and corresponding clinical pathological information of 614 KIRC samples were downloaded from the TCGA database. R software (version 4.1.2) was used for statistical analysis. The “edgeR” package was utilized to screen for DEGs, with criteria set at an adjusted P-value < 0.05 and |log2 Fold Change (FC)|> 1.
Construction of a prognostic model based on integrated machine learning methods
To establish a consensus of PCD-related genes with high accuracy and stability, we integrated 10 machine learning algorithms and 101 algorithm combinations. These algorithms include Random Survival Forest (RSF), Elastic Net (Enet), Lasso, Ridge, Stepwise Cox, CoxBoost, Partial Least Squares Regression (plsRcox), Supervised Principal Component (SuperPC), Generalized Boosted Regression Modeling (GBM), and Support Vector Machine for Survival (survival-SVM),were obtained from R Project (https://www.r-project.org/) . The steps for generating the feature signature include: (a) constructing predictive models in the TCGA-KIRC cohort using 101 algorithm combinations and leave-one-out cross-validation (LOOCV); (b) further cross-validation of all models using GSE22541; (c) calculating the Harrell’s Concordance Index (C-index) for each model, with models having an average C-index or test set C-index > 0.7 considered excellent. According to the descriptions in previous literature26,27, we stratified KIRC patients into high and low risk groups based on the median scores of their respective cohorts.
Construction and evaluation of nomograms
To validate the value of different PCD patterns as independent prognostic indicators for KIRC patients, univariate and multivariate Cox regression analyses were performed. Prognostic nomograms were developed using the R packages “rms” and “replot” based on the TCGA-KIRC and GSE22541 cohorts, and their performance was evaluated through the Receiver Operating Characteristic (ROC) curves.
Immune infiltration analysis
Comprehensive immune cell infiltration analysis of the TCGA-KIRC dataset samples was performed using the IOBR package (https://github.com/IOBR/IOBR) in conjunction with the CIBERSORT algorithm. Pearson correlation analysis was employed to explore the association between differentially expressed genes and specific immune cell subpopulations. Functional enrichment analysis of immune cells was conducted using the immunogenomic "immune.gmt".
Prediction of immunotherapy response
Tumor immune dysfunction and exclusion (TIDE) is a computational framework (http://tide.dfci.harvard.edu/) that assesses the potential for tumor immune evasion by evaluating the gene expression profiles of cancer samples. The TIDE score, composed of immune dysfunction and immune exclusion components, serves as an alternative biomarker for predicting the response to immune checkpoint blockade across various cancer types. The list of immune checkpoint genes (ICGs) is derived from prior research28.
Somatic mutation analysis
Somatic mutation data downloaded from the TCGA database were analyzed using the “maftools” package in R. Tumor mutational burden (TMB) was calculated as the number of non-synonymous somatic mutations per megabase in cancer samples.
GSEA
GSEA software version 3.0 was obtained from the official GSEA website. Samples were stratified into high expression (> 50%) and low expression (< 50%) groups based on the expression of hub genes. The gene set c2.cp.kegg.v7.4.symbols.gmt was downloaded for analyzing biological pathways and molecular mechanisms29. Through GSEA analysis, with a minimum gene set size of 5 and a maximum of 5000, and performing a thousand simulation samplings, pathways with P < 0.05 and normalized enrichment score thresholds were selected and categorized for visualization.
TME scoring
The ESTIMATE algorithm was used to analyze the infiltration levels of immune cells and stromal cells in KIRC, with data sourced from the TCGA database. Stromal scores, immune scores, and composite scores were calculated.
Drug sensitivity analysis
The response of KIRC patients to antitumor drugs was predicted based on the Genomics of Drug Sensitivity in Cancer (GDSC) database, reflected by the maximum half-maximal inhibitory concentration (IC50). The “ggplot2” R package was used to analyze and display the differences in IC50 among different groups of KIRC patients.
Docking validation of drug small molecule components and core common target molecules
The 3D structures (.pdb format files) of core common target molecules were downloaded from the PDB database (https://www.rcsb.org/). The structures of small molecule drugs (.sdf format files) were collected and analyzed from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). Molecular docking was performed using the CB-Dock2 online database (https://cadd.labshare.cn/cb-dock2/php/blinddock.php) by uploading the small molecule compounds and target proteins into the system. A higher absolute value of the Vina score indicates a stronger binding capacity of the ligand molecule to the receptor protein. The specific parameter setting principle is to use the coordinates of the compound originally bound in the pocket of the protein target as the center for lattice construction.
Results
Preliminary screening of PCD of KIRC patient
Our research workflow is depicted in Fig. 1. Initially, we collected essential regulatory genes encompassing 18 modes of PCD from literature, totaling 1964 genes20 (Fig. 2A, Supplementary Tables S1). Subsequently, differential analysis was conducted on the TCGA-KIRC cohort to identify 12,788 genes that exhibit differential expression between normal and tumor tissues (Fig. 2B). Intersections were then taken between genes related to the 18 PCD modes and differentially expressed genes. It was found that there were 157 intersecting genes with Apoptosis, 120 with Anoikis, 65 with Autophagy, 55 with LDCD, 26 with Necroptosis, 22 with Ferroptosis, 20 with Pyroptosis, 16 with Paraptosis, 14 with Immunogenic cell death, 8 with NETosis, 6 with Cuproptosis, 4 with Entosis, 3 with Entotic cell death, 3 with Methuosis, 2 with Netotic cell death, 1 with Alkaliptosis, and 1 with Oxeiptosis, while no intersecting genes were found with Parthanatos (Fig. 2C). We included 11 types of cell death modes with more than 5 intersecting genes and, through univariate COX regression analysis, found that most PCD modes were associated with risk factors, whereas genes related to LDCD and Cuproptosis were predominantly protective factors (Fig. 3A–K).

A graphic abstract of this study.

Preliminary screening of PCD of KIRC patient. (A) Essential regulatory genes for 18 modes of PCD. (B) 12,788 DEGs between normal and tumor tissues in the TCGA-KIRC cohort. (C) Intersections of 18 PCD modes with differentially expressed genes in the TCGA-KIRC cohort.

Univariate COX regression analysis results for 11 PCD modes. (A) Anoikis, (B) Apoptosis, (C) Autophagy, (D) LDCD, (E) Ferroptosis, (F) Necroptosis, (G) Immunogenic cell death, (H) NETosis, (I) Cuproptosis, (J) Paraptosis, (K) Pyroptosis.
101 machine learning combinations unveil key PCD patterns in KIRC
Our study successfully constructed a consensus riskscore model based on combinations of 101 algorithms and calculated the C-index for 11 cell death patterns across all cohorts in the TCGA-KIRC and GSE22541 datasets to evaluate the predictive efficacy of the model. The results indicated that the models for Anoikis, ferroptosis, LDCD, and autophagy demonstrated the highest average C-index or excellent performance, with a C-index exceeding 0.7 in the test set (Fig. 4A, B, Supplementary Figures S1A-I). Furthermore, we assessed the diagnostic efficacy of PCD in the cohorts by calculating the area under the ROC curve, where the Anoikis and LDCD models were ranked at the top (Fig. 4C and E, Supplementary Figures S1J-K). Survival analysis revealed that the high-risk groups for both the Anoikis and LDCD models had significantly adverse prognoses (Fig. 4D and F). Through multivariate analysis and calculation of the C-index for clinical characteristics, we found that the Anoikis riskscore model surpassed traditional clinical characteristics such as age, gender, tumor grade, and clinical stage in predictive power (Fig. 4G). Although the C-index of the LDCD riskscore model was slightly lower than that of clinical staging, its predictive stability was more pronounced (Fig. 4H). To enhance the clinical application value of the model further, we integrated the riskscore with clinical factors such as gender and age to develop nomograms as a novel prognostic model, where the Anoikis nomogram model achieved a predictive performance of 0.781, and the LDCD nomogram model achieved a predictive performance of 0.770 (Fig. 5A-D). These findings provide a new perspective and tools for the prognostic assessment of KIRC patients.

101 Machine learning combinations unveil key patterns of PCD in KIRC. (A-B) Model construction of TCGA-KIRC and GSE2254. (C-D) ROC curves and survival analysis for anoikis. (E–F) ROC curves and survival analysis for LDCD. (G-H) Predictive ability and comparative diagram of anoikis and LDCD.

Nomogram models. (A, B) Anoikis. (C, D) LDCD.

Immune cell survival analysis and phenotyping. Immune cell survival analysis is presented for the following panels: (A) Tregs, (B) Tfh, (C) CD8 T cells, (D) CD4 memory resting T cells, (E) CD4 memory activated T cells, (F) Plasma cells, (G) Monocytes, (H) Resting mast cells, (I) Activated mast cells, (J) Macrophages M2, (K) Macrophages M0, and (L) Resting dendritic cells. Immune cell phenotyping for (M) Anoikis and (N) LDCD.
Analysis of the association between immune cell infiltration and typing in KIRC patients
This study conducted an in-depth exploration of the relationship between the immune cell infiltration status in the TCGA-KIRC cohort and patient clinical outcomes. The results indicated that higher infiltration levels of T cells CD4 memory resting, monocytes, mast cells resting, macrophages M2, and dendritic cells resting were associated with a positive improvement in overall patient survival prognosis. Conversely, higher infiltration levels of plasma cells, T cells CD8, T cells CD4 memory activated, Tfh, T cells regulatory (Tregs), macrophages M0, macrophages M2, dendritic cells resting, and mast cells activated were correlated with a worse prognosis for the patients. We further analyzed that within the Anoikis and LDCD death patterns, the low-risk group exhibited higher levels of inflammatory immune cell subtypes compared to the high-risk group, with the expression levels of inflammatory immune cell subtypes being higher in the low-risk group than in the high-risk group (Fig. 6).
Immune cell subtypes and immune checkpoint expression in anoikis and LDCD
Our study reveals that in the context of Anoikis, the high-risk group exhibits heightened expression of specific immune cell subsets. Specifically, T cells CD4 memory resting, T cells CD4 memory activated, Tfh, Tregs, Macrophages M0, and activated mast cells are more prevalent in these patients. Concurrently, resting dendritic cells and resting mast cells show relatively lower expression levels in the high-risk group (Fig. 7A). Furthermore, in the case of LDCD, we observed that plasma cells, CD8 T cells, Tfh, and Tregs have higher expression levels in the high-risk group. In contrast, CD4 memory resting T cells, macrophages M2, resting mast cells, and activated mast cells exhibit lower expression in the high-risk group (Fig. 7B). To further validate these findings, we analyzed the correlation of immune cells in the two types of PCD using various software algorithms. Figure 7C, D display the results of these analyses, providing a quantitative perspective on the expression patterns of immune cell subsets and potentially revealing the interplay between different immune cell subsets and their impact on the tumor microenvironment. The results of immune cell functional analysis indicate that the expression of immune checkpoints is generally higher in the high-risk group, both in Anoikis and LDCD (Fig. 7E, F).

Immune cell subtypes and immune checkpoint expression. (A, B) Analysis of immune cell infiltration for Anoikis and LDCD. (C, D) Analysis of immune cell infiltration using diverse software algorithms. (E, F) Functional enrichment analysis of immune cells.
Molecular characteristics and immune escape mechanisms of anoikis and LDCD
Figure 8A–B’s heatmaps display the clinical information of patients under the two modes of PCD. Within the TCGA-KIRC cohort, we delved into the key biological differences among various PCD patterns. In the somatic mutation analysis of the Anoikis, we observed that the overall mutation rate in the high-risk group (Fig. 8C, 53.21%) was significantly higher than that in the low-risk group (Fig. 8D, 41.09%). Notably, the mutation rates of the TTN and MUC16 genes were significantly higher in both risk groups. Additionally, the the TMB in the high-risk group was higher than that in the low-risk group (Fig. 8E, F). In LDCD, although the overall mutation rate in the high-risk group (Fig. 8G, 82.66%) was slightly higher than in the low-risk group (Fig. 8H, 81.62%), there was no significant difference in TMB between the two (Fig. 8I, J). Furthermore, we analyzed the expression relationship between the two PCD patterns and immune checkpoint-related genes. In Anoikis, the expression levels of most immune checkpoint-related genes were significantly elevated in the high-risk group (Fig. 8K). Similarly, in LDCD, immune checkpoint-related genes were also highly expressed in the high-risk group (Fig. 8L). To gain a deeper understanding of the immune escape mechanisms relevant to immunotherapy, we conducted further analyses. The results showed that in Anoikis, the high-risk group had higher microsatellite instability, TIDE scores, immune dysfunction, responder status, CD8+ T cells, CD274 (also PD-L1) expression, and TAM M2 phenotype, while immune exclusion and cancer-associated fibroblasts (CAF) were not statistically significant in high-risk group patients (Fig. 9A–I). In LDCD, a similar pattern was observed, with the high-risk group also exhibiting higher microsatellite instability, TIDE scores, immune dysfunction, responder status, CD8+ T cells, CD274 expression, and TAM M2 phenotype, while immune exclusion, CAF, and CD274 expression were not statistically significant in high-risk group patients (Fig. 9J–R).

Molecular characteristics and immune escape mechanisms. (A, B) Clinical information of patients under Anoikis and LDCD. (C, D) Somatic mutation analysis for Anoikis. (E, F) TMB analysis for Anoikis. (G, H) Somatic mutation analysis for LDCD. (I, J) TMB analysis for LDCD. (K, L) Expression levels of immune checkpoint-related genes in Anoikis and LDCD.

Analysis of immune evasion correlation. (A–I) Analysis of immune checkpoint associations in high and low-risk Anoikis groups: (A) MSI, (B) TIDE, (C) Dysfunction, (D) Exclusion, (E) CAF, (F) Responder, (G) CD8, (H) CD274, (I) TAM M2. (J–R) Analysis of immune checkpoint associations in high and low-risk LDCD groups: (J) MSI, (K) TIDE, (L) Dysfunction, (M) Exclusion, (N) CAF, (O) Responder, (P) CD8, (Q) CD274, (R) TAM M2.
GSEA enrichment analysis and tumor microenvironment assessment of Anoikis and LDCD
We conducted further GSEA for two key modes of PCD in KIRC. In the context of Anoikis, Gene Ontology (GO) analysis results highlighted significant enrichment of immune-related pathways in the high-risk group (Fig. 10A). Concurrently, gene expression in the low-risk group was significantly enriched in biological processes related to the renal system and cell polarization (Supplementary Figures S2A). Subsequent Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed significant enrichment in immune response and cell signaling pathways for the high-risk group (Supplementary Figures S2B), while the low-risk group showed significant enrichment in those related to metabolic processes pathway (Supplementary Figures S2C). In the scenario of LDCD, GO analysis results disclosed significant enrichment in processes related to spliceosome assembly and function for the high-risk group (Supplementary Figures S2D). The low-risk group demonstrated significant enrichment in processes related to ion transport, renal function, and metabolism (Supplementary Figures S2E). KEGG results indicated no significant enrichment for the high-risk group, while the low-risk group continued to show enrichment in the processes related to drug metabolism, xenobiotic detoxification, and amino acid metabolism (Supplementary Figures S2F). By employing the ESTIMATE algorithm within the R package ‘estimate’, we estimated the proportions of immune and stromal components in the tumor microenvironment for high- and low-risk groups in both PCD patterns, obtaining three scores: ImmuneScore, StromalScore, and ESTIMATEScore. The high-risk group correlated positively with these scores, indicating an association with immune and stromal components (Fig. 10B, C). Given the significant similarities exhibited by Anoikis and LDCD in immune-related analyses, enrichment analyses, and immune stromal components, we conducted an integrated analysis of Anoikis-related genes, LDCD-related genes, and DEGs in KIRC patients, identifying two key intersecting genes, HMOX1 and PIK3CG (Fig. 10D), which may play a crucial role in the immune response and therapeutic response in KIRC.

GSEA and assessment of the tumor microenvironment. (A) GO analysis of high-risk groups for Anoikis. (B, C) Ratios of immune stroma components in the tumor microenvironment among high and low-risk groups for Anoikis and LDCD. (D) Intersection of Anoikis, LDCD, and DEGs in KIRC patients.
Drug sensitivity analysis of Anoikis and LDCD
Given that the prognosis of the high-risk group is significantly poorer than that of the low-risk group, we conducted a drug analysis for both the low- and high-risk groups of the two types of PCD to predict drugs that may exhibit higher sensitivity in the high-risk group. The results indicated that in the high-risk group of Anoikis, dihydrorotenone, OF-1, ibrutinib, SB505124, and P22077 showed greater sensitivity (Fig. 11A–E). In the high-risk group of LDCD, only SB505124 demonstrated higher sensitivity (Fig. 11F). Interestingly, we found that SB505124 exhibited high sensitivity in the high-risk groups of both types of PCD.

Molecular docking verification of HMOX1 and PIK3CG with related drug small molecule components. (A–E) Anoikis exhibits sensitivity to the drugs dihydrorotenone, OF-1, ibrutinib, SB505124, and P22077. (F) LDCD shows sensitivity to SB505124. (G–J) Schematic diagrams of molecular docking (black circles represent binding pockets, and dashed lines of different colors represent potential weak interactions between ligands and amino acid sites).
Molecular docking verification of HMOX1 and PIK3CG with related drug small molecule components
As HMOX1 and PIK3CG are key genes in the two types of PCD, we performed molecular docking analysis on them. Through literature review, it was found that Carnosol exerts its antioxidant and microvascular endothelial cell-protective functions by enhancing the expression of HMOX1 and activating the Nrf2 signaling pathway30. Therefore, we verified the binding capacity of Carnosol with HMOX1, confirming that Carnosol can form a stable binding with the active site of HMOX1 (Fig. 11G, H). Concurrently, we explored the molecular targeting strategy for PIK3CG. According to the literature, a research team developed a PROTAC bispecific molecule that can specifically degrade PIK3CG, thereby blocking the PI3Kγ-Akt signaling pathway31. Our molecular docking verification showed that PIK3CG can form a stable binding with the active site of the PROTAC molecule (Fig. 11I, J), providing a novel strategy for targeting PIK3CG in therapy.
Discussion
KIRC is a prototypical tumor with high immune infiltration, and its heterogeneous response to treatment has long been a significant challenge for clinical precision diagnosis and treatment10. In this study, we systematically dissected the synergistic regulatory network of anoikis and LDCD in KIRC for the first time by integrating machine learning and multi-omics analysis. Our findings elucidate the molecular mechanisms by which these two modes of PCD mediate treatment resistance through remodeling of the immunosuppressive microenvironment, providing a novel theoretical framework and potential therapeutic strategies to overcome resistance to immune checkpoint inhibitors .
We confirmed that the high-risk phenotype of anoikis is significantly associated with poor prognosis in KIRC patients, which contrasts sharply with the traditional view of anoikis as a metastasis-inhibiting mechanism32. Previous studies have suggested that KIRC may achieve immune evasion by constructing a multi-dimensional anoikis resistance network. At the level of immune checkpoint regulation, the mechanism by which the FXa-PAR-2 signaling axis activates the NF-κB pathway to induce PD-L1 transcriptional upregulation has been reported in hepatocellular carcinoma33, consistent with the elevated PD-L1 expression observed in the high-risk anoikis group in our study. In terms of the immunosuppressive cell network, the SPP1-mediated recruitment of MDSCs in hepatocellular carcinoma suggests that KIRC may establish an immunosuppressive barrier through a similar pathway34. At the metabolic-immune intersection, the metabolic reprogramming mechanism mediated by glutamate dehydrogenase 1 and ribosomal S6 kinase 2 in lung cancer offers new insights into the inhibition of CD8 + T-cell infiltration in KIRC35. Notably, our study breaks through the paradigm of considering PCD as an isolated mechanism by revealing for the first time a correlation between anoikis and LDCD in KIRC, suggesting that these two processes may amplify immunosuppressive effects through synergistic action.
LDCD is a form of PCD triggered by lysosomal membrane permeabilization (LMP), with its core mechanism involving the release of lysosomal hydrolases that disrupt intracellular homeostasis36. In the regulation of the tumor immune microenvironment, LDCD exhibits a dual mode of action. On one hand, LMP-mediated release of lysosomal content induces the exposure of damage-associated molecular patterns (DAMPs), which activate innate immune responses to facilitate the clearance of dying cells37,38,39. On the other hand, tumor cells can hijack the LDCD pathway to achieve immune evasion through multiple mechanisms. Specifically, abnormal release of lysosomal components such as cathepsin D can activate the caspase-8 signaling axis, inhibiting immune cell activation through a cascade of reactions40. Tumor cells also reshape the lysosomal acidic environment and membrane stability to evade LMP-mediated death while suppressing the metabolic reprogramming capacity of immune cells, leading to terminal differentiation and exhaustion of plasma cells and lymphocytes41,42. The spatiotemporal-specific release of lysosomal content may regulate the immunogenicity of cell death, and when this process is disrupted, it not only fails to effectively recruit immune cells but also promotes immune tolerance by inhibiting antigen presentation. Previous studies have shown that the expression levels of LDCD-related genes are positively correlated with the degree of immune cell infiltration, particularly B cells and Treg cells, providing new molecular targets for elucidating the formation of the immunosuppressive microenvironment in KIRC43.
Based on these mechanisms, this study innovatively proposes a combination therapy strategy targeting the PCD synergistic network. Drug screening results show that the TGF-β receptor inhibitor SB505124 can simultaneously suppress the high-risk phenotypes of anoikis and LDCD. SB505124 blocks the TGF-β signaling pathway, thereby effectively inhibiting cell proliferation, differentiation, and migration44. Although no studies have directly explored the specific role of SB505124 in KIRC treatment to date, TGF-β signaling has been identified as a key regulator of KIRC invasiveness and metastatic potential45,46. Considering the central role of the TGF-β pathway in KIRC malignancy46 and T-cell regulation47, the combination of SB505124 with PD-1 inhibitors is expected to reverse immunosuppression and amplify PCD effects.
In addition, we found that HMOX1 and PIK3CG are both expressed in anoikis-, LDCD-related genes, and differentially upregulated genes in KIRC patients. This suggests that anoikis and LDCD may enhance the expression of HMOX1 and PIK3CG, thereby affecting Tfh infiltration and HLA-E expression to shape the immunotherapy response in KIRC patients. HMOX1 has been confirmed to play a regulatory role in the immunotherapy of advanced renal cell carcinoma48, consistent with our findings. Moreover, through literature review, we found that Carnosol can enhance HMOX1 expression and activate the Nrf2 signaling pathway, exerting antioxidant and protective effects on microvascular endothelial cells30. Our experiments further verified the binding capacity of Carnosol to the active sites of HMOX1, confirming its stability. Although there is currently a lack of literature directly studying the impact of PIK3CG on KIRC immunotherapy, as a key component of the PI3K/Akt/mTOR signaling pathway, its potential role in KIRC treatment should not be overlooked, especially since mTOR inhibitors have shown significant efficacy in KIRC treatment49. Furthermore, a research team has developed a PROTAC heterobifunctional molecule that can specifically degrade PIK3CG and block the PI3Kɣ-Akt signaling pathway50. Our further molecular docking analysis revealed stable binding between the active sites of PIK3CG and the PROTAC molecule. Through additional molecular docking analysis, we validated the binding affinity between the active sites of HMOX1 and PIK3CG and potential drug molecules, providing important information for the development of new therapeutic strategies.
It is noteworthy that although KIRC is a tumor resistant to conventional radiotherapy, recent studies have shown that radioresistance can be overcome by high doses delivered through stereotactic body radiotherapy51. Evidence that radiotherapy can stimulate local52 and distant antitumor immunity (a phenomenon known as the “abscopal effect”53) has paved the way for combining radiotherapy with emerging immunotherapies, such as anti-PD-154. In terms of programmed cell death, radiotherapy can induce pyroptosis in tumor cells, a potent form of PCD that activates the immune response55. For example, research teams have confirmed that radiotherapy can induce pyroptosis in certain types of tumor cells, leading to the release of IL-1β and IL-18, thereby exerting tumor-suppressive functions and inducing antitumor immune responses56. In contrast, immune checkpoint inhibitors target dysfunctional immune systems to induce CD8 + T cells to kill cancer cells57, while CD8 + T cells inhibit tumor growth through the induction of ferroptosis, necrosis, and pyroptosis58,59. Therefore, both immune checkpoint inhibitor therapy and SBRT have their respective advantages in inducing programmed cell death. Immune checkpoint inhibitors induce multiple modes of programmed cell death through systemic immune responses and direct damage to tumor cells, while radiotherapy primarily activates the immune response through local high-energy radiation damage and induction of pyroptosis, indicating that radiotherapy can serve as a potential partner for immunotherapy. Moreover, the identification of novel immune targets will improve the efficacy and long-term response rates of combination therapies. As the number of RCC immunotherapy trials increases and radiotherapy techniques advance, new opportunities for RCC management will emerge.
In summary, this study overcomes the limitation of focusing solely on a single mode of PCD by revealing the interaction between anoikis and LDCD for the first time and demonstrating how they collectively shape the immunotherapy response in patients with KIRC. Specifically, we found that anoikis and LDCD may enhance the expression of HMOX1 and PIK3CG, thereby influencing the infiltration of Tfh cells and the expression of HLA-E, which together shape the immunotherapy responsiveness in KIRC patients. Additionally, we discovered that SB505124 exhibits significant sensitivity in the high-risk groups of both PCD modes. We also confirmed stable binding interactions between Carnosol and HMOX1, as well as between PROTAC and PIK3CG. Our study underscores the necessity of considering the interplay among multiple PCD pathways in the therapeutic strategies for KIRC, which is crucial for identifying key target drugs and developing combination therapies, with the hope of bringing more precise and effective treatment options to KIRC patients.
Conclusion
Our study reveals that Anoikis and LDCD may diminish the immune response in KIRC patients by enhancing the expression of HMOX1 and PIK3CG. Furthermore, SB505124, Carnosol, and PROTAC have demonstrated significant potential as candidate immunotherapeutic agents.
Limitations
While bioinformatics analysis provides preliminary insights and hypotheses, relying solely on these analyses may not fully reveal the complexity of these biological phenomena. Therefore, it is planned to translate these preliminary findings into laboratory experimental validations in future research, thereby providing a solid foundation for the development of more effective therapeutic strategies.
Data availability
RNA sequencing data and corresponding clinical pathological information of 614 KIRC samples were obtained from the TCGA database (portal.gdc.cancer.gov/projects/TCGA-KIRC). And the clinical pathological information and microarray expression analysis data of the KIRC cohort GSE22541 were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/gds).
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249 (2021).
Pontes, O. et al. Renal cell carcinoma therapy: current and new drug candidates. Drug Discov. Today 27(1), 304–314 (2022).
Wang, Y. Z. & Hu, Y. Age-period-cohort analysis on kidney cancer mortality trend in China, 1992–2016. Zhonghua Liu Xing Bing Xue Za Zhi 42(3), 508–512 (2021).
Ji, Y. T. et al. Comparison of the latest cancer statistics, cancer epidemic trends and determinants between China and the United States. Zhonghua Zhong Liu Za Zhi 46(7), 646–656 (2024).
Moch, H. et al. The 2022 world health organization classification of tumours of the urinary system and male genital organs-part a: renal, penile, and testicular tumours. Eur. Urol. 82(5), 458–468 (2022).
Li, P. et al. Survival among patients with advanced renal cell carcinoma in the pretargeted versus targeted therapy eras. Cancer Med. 5(2), 169–181 (2016).
Choueiri, T. K. et al. Overall survival with adjuvant pembrolizumab in renal-cell carcinoma. N. Engl. J. Med. 390(15), 1359–1371 (2024).
Luo, D. et al. Targeted chemoradiotherapy of prostate cancer using gold nanoclusters with protease activatable monomethyl auristatin E. ACS Appl. Mater. Interfaces 14(13), 14916–14927 (2022).
Huang, C. P. et al. ASC-J9(R) increases the bladder cancer chemotherapy efficacy via altering the androgen receptor (AR) and NF-kappaB survival signals. J. Exp. Clin. Cancer Res. 38(1), 275 (2019).
Vuong, L. et al. Tumor microenvironment dynamics in clear-cell renal cell carcinoma. Cancer Discov. 9(10), 1349–1357 (2019).
Pitt, J. M. et al. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 27(8), 1482–1492 (2016).
Lai, Y. et al. The tumour microenvironment and metabolism in renal cell carcinoma targeted or immune therapy. J. Cell Physiol. 236(3), 1616–1627 (2021).
Ricketts, C. J. et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 43(4), 113063 (2024).
Braun, D. A. et al. Beyond conventional immune-checkpoint inhibition—novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 18(4), 199–214 (2021).
Dai, S. et al. Intratumoral CXCL13(+)CD8(+)T cell infiltration determines poor clinical outcomes and immunoevasive contexture in patients with clear cell renal cell carcinoma. J. Immunother. Cancer 9, 2 (2021).
Golkaram, M. et al. Spatiotemporal evolution of the clear cell renal cell carcinoma microenvironment links intra-tumoral heterogeneity to immune escape. Genome Med. 14(1), 143 (2022).
Wu, X. et al. CD8(+) T cell-based molecular classification with heterogeneous immunogenomic landscapes and clinical significance of clear cell renal cell carcinoma. Front. Immunol. 12, 745945 (2021).
Tang, D. et al. The molecular machinery of regulated cell death. Cell Res. 29(5), 347–364 (2019).
Newton, K. et al. Cell death. Cell 187(2), 235–256 (2024).
Qin, H. et al. Integrated machine learning survival framework develops a prognostic model based on inter-crosstalk definition of mitochondrial function and cell death patterns in a large multicenter cohort for lower-grade glioma. J. Transl. Med. 21(1), 588 (2023).
Zou, Y. et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int. J. Surg. 107, 106936 (2022).
Luo, Y., Liu, L. & Zhang, C. Identification and analysis of diverse cell death patterns in diabetic kidney disease using microarray-based transcriptome profiling and single-nucleus RNA sequencing. Comput. Biol. Med. 169, 107780 (2024).
Liu, J. et al. Programmed cell death tunes tumor immunity. Front. Immunol. 13, 847345 (2022).
Guo, J. S. et al. Cuproptosis-related 4-gene risk model for predicting immunotherapy drug response and prognosis of kidney renal clear cell carcinoma. Chin. Med. Sci. J. 38(3), 191–205 (2023).
Dong, P. et al. A model based on immunogenic cell death-related genes predicts prognosis and response to immunotherapy in kidney renal clear cell carcinoma. Transl. Cancer Res. 13(1), 249–267 (2024).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13(1), 816 (2022).
Feng, S. et al. Integrated clinical characteristics and omics analysis identifies a ferroptosis and iron-metabolism-related lncRNA signature for predicting prognosis and therapeutic responses in ovarian cancer. J. Ovarian Res. 15(1), 10 (2022).
Hu, F. F. et al. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinform. 22, 3 (2021).
Kanehisa, M. et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44(D1), D457–D462 (2016).
Li, X. et al. Carnosol as a Nrf2 activator improves endothelial barrier function through antioxidative mechanisms. Int. J. Mol. Sci. 20, 4 (2019).
Kelly, L. M. et al. Targeting a lineage-specific PI3Kɣ-Akt signaling module in acute myeloid leukemia using a heterobifunctional degrader molecule. Nat. Cancer 2024, 56 (2024).
Eble, J. A. & Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 36(3), 171–198 (2019).
Li, X. et al. FXa-mediated PAR-2 promotes the efficacy of immunotherapy for hepatocellular carcinoma through immune escape and anoikis resistance by inducing PD-L1 transcription. J. Immunother. Cancer 12, 7 (2024).
Gonzalez, M. J. et al. Influence of hydroclimatic conditions and anthropogenic activities on the water quality of a floodplain lake (Argentina) during a warm season. Environ. Sci. Pollut. Res. Int. 31(36), 49330–49341 (2024).
Kang, J. et al. EGFR-phosphorylated GDH1 harmonizes with RSK2 to drive CREB activation and tumor metastasis in EGFR-activated lung cancer. Cell Rep. 41(11), 111827 (2022).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25(3), 486–541 (2018).
Ma, M., Jiang, W. & Zhou, R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity 57(4), 752–771 (2024).
Zhu, M. Y. et al. LW-213 induces immunogenic tumor cell death via ER stress mediated by lysosomal TRPML1. Cancer Lett. 577, 216435 (2023).
Sarhan, M. et al. Immunological consequences of kidney cell death. Cell Death Dis. 9(2), 114 (2018).
Conus, S. et al. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J. Exp. Med. 205(3), 685–698 (2008).
Yuseff, M. I. et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity 35(3), 361–374 (2011).
Kimmey, J. M. & Stallings, C. L. Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol. Med. 22(12), 1060–1076 (2016).
He, S. et al. Molecular characteristics and prognostic significances of lysosomal-dependent cell death in kidney renal clear cell carcinoma. Aging (Albany N.Y.) 16(5), 4862–4888 (2024).
Dacosta-Byfield, S. et al. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 65(3), 744–752 (2004).
Fang, Z. et al. GABPA-activated TGFBR2 transcription inhibits aggressiveness but is epigenetically erased by oncometabolites in renal cell carcinoma. J. Exp. Clin. Cancer Res. 41(1), 173 (2022).
Cui, H. et al. The SWI/SNF chromatin remodeling factor DPF3 regulates metastasis of ccRCC by modulating TGF-beta signaling. Nat. Commun. 13(1), 4680 (2022).
Chen, W. TGF-beta regulation of T cells. Annu. Rev. Immunol. 41, 483–512 (2023).
Xu, X. et al. HMOX1 pathway signature predicts clinical benefit from immunotherapy plus tyrosine kinase inhibitor therapy in advanced renal cell carcinoma. Cancer Med. 12(9), 10512–10525 (2023).
Makhov, P. et al. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol. Cancer Ther. 17(7), 1355–1364 (2018).
Kelly, L. M. et al. Targeting a lineage-specific PI3Kɣ-Akt signaling module in acute myeloid leukemia using a heterobifunctional degrader molecule. Nat. Cancer 5(7), 1082–1101 (2024).
Sun, X. et al. Combination with stereotactic body radiotherapy offers a promising strategy to overcome resistance to immunotherapy in advanced renal cell cancer. J. Oncol. 2019, 1483406 (2019).
Siva, S. et al. Radiotherapy for renal cell carcinoma: renaissance of an overlooked approach. Nat. Rev. Urol. 14(9), 549–563 (2017).
Walle, T. et al. Radiation effects on antitumor immune responses: current perspectives and challenges. Ther. Adv. Med. Oncol. 10, 1758834017742575 (2018).
Spaas, M. et al. Checkpoint inhibitors in combination with stereotactic body radiotherapy in patients with advanced solid tumors: The CHEERS phase 2 randomized clinical trial. JAMA Oncol. 9(9), 1205–1213 (2023).
Wang, Y. et al. Ferroptosis, necroptosis, and pyroptosis in cancer: Crucial cell death types in radiotherapy and post-radiotherapy immune activation. Radiother. Oncol. 184, 109689 (2023).
Tan, Y. et al. Pyroptosis: a new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 40(1), 153 (2021).
Carlino, M. S., Larkin, J. & Long, G. V. Immune checkpoint inhibitors in melanoma. Lancet 398(10304), 1002–1014 (2021).
Tang, R. et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 13(1), 110 (2020).
Yu, L. et al. Targeting novel regulated cell death: ferroptosis, pyroptosis and necroptosis in anti-PD-1/PD-L1 cancer immunotherapy. Cell Prolif 57(8), e13644 (2024).
Author information
Authors and Affiliations
Contributions
J.Y. and F.O. conceived and supervised the entire project. Y.P. and Q.C. wrote the code and drafted the manuscript. L.X. Zeng, K.W. and D.L. processed the data and analyzed the results. L.X.Zeng taked charge of the creation of figures and the layout of grouped figures. Q.G. and L.Q. Zhou. improved the algorithms and reviewed the manuscript. All authors discussed, revised, and proofread the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ou, F., Pan, Y., Chen, Q. et al. Integrating machine learning and multi-omics analysis to unveil key programmed cell death patterns and immunotherapy targets in kidney renal clear cell carcinoma. Sci Rep 15, 18403 (2025). https://doi.org/10.1038/s41598-025-00759-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-00759-z
