
Abstract
Persistent pulmonary hypertension of the newborn (PPHN) represents a life-threatening cardiopulmonary condition characterized by hypoxia-driven pulmonary vascular remodeling. While transcription factor EB (TFEB), a master regulator of cellular adaptation to hypoxia, has been implicated in vascular pathologies, its mechanistic role in PPHN remains undefined. This study elucidates the molecular interplay of TFEB in hypoxia-induced PPHN pathogenesis. Fetal rat models of hypoxia-induced PPHN, including untreated hypoxic models and hypoxic models treated with the TFEB inhibitor Eltrombopag (EO), as well as a hypoxia-induced human pulmonary artery endothelial cell (HPAEC) model, were established. Multimodal assessments, including histopathology, qRT-PCR, JC-1 staining, immunofluorescence, flow cytometry, and Western blotting, were employed to evaluate the effects of TFEB on mitophagy and NLRP3 inflammasome. In the hypoxia group, significant thickening of the pulmonary arterioles and right ventricular wall was observed. Immunostaining revealed a significant increase in the relative staining density of TFEB-positive, NLRP3-positive, and LC3-positive cells, alongside elevated expression of mitophagy-proteins and NLRP3 inflammasome-related proteins. TFEB inhibition downregulated the expression of PINK1, TOMM20, COX IV, P62, and LC3II/I ratio, impairing mitophagy, while upregulating eNOS, NLRP3, and GSDMD, thereby enhancing NLRP3 activation and pyroptosis. In the EO group, fetal rats exhibited more pronounced pulmonary arteriole thickening, intensified fluorescence signals for NLRP3, caspase-1, and GSDMD, reduced mitophagy-related protein expression, and further elevated NLRP3 inflammasome-related protein and GSDMD expression. TFEB exerts a protective effect in PPHN by inhibiting NLRP3 inflammasome activation through PINK1/Parkin-mediated mitophagy, highlighting TFEB’s potential as a therapeutic target for hypoxia-induced PPHN.
Similar content being viewed by others


Introduction
Persistent pulmonary hypertension of the newborn (PPHN) is a severe clinical syndrome characterized by increased pulmonary vascular resistance secondary to refractory and severe hypoxemia1. This condition typically results from a multifactorial etiology, with oxidative stress being widely recognized as a critical pathophysiological contributor to PPHN2. Specifically, mitochondrial oxidative stress exacerbates vascular injury in pulmonary hypertension, inducing vascular remodeling3,4,5. Despite these established associations, the precise mechanistic relationship between these pathological changes and the development of PPHN remains poorly understood, highlighting the need for further mechanistic investigation.
Transcription factor EB (TFEB) is a central regulator of autophagy and the lysosomal-nuclear signaling pathway6,7. Elevated TFEB expression can significantly reduce the inflammatory reaction in lung tissue and epithelial cells, while enhancing autophagy levels and soluble protein production8. Mitophagy, mediated by the PTEN-induced putative kinase 1 (PINK1)/Parkin RBR E3 ubiquitin ligase (Parkin) pathway, represents a critical self-recovery mechanism with essential physiological functions9. Under conditions of cellular stress or adverse stimuli, PINK1 triggers the phosphorylation of Parkin and initiates mitophagy. This process enables cells to restore immune responses and cellular homeostasis while efficiently eliminating damaged mitochondria10,11. Emerging evidence suggests that TFEB overexpression can mitigate lipopolysaccharide-induced acute lung injury through the regulation of mitophagy12. Furthermore, TFEB activation has been shown to promote mitophagy via the Parkin pathway, alleviating oxidative stress-induced intestinal barrier dysfunction and mitochondrial damage13. While TFEB has been extensively studied in various pathological conditions, including pneumonia, sepsis, atherosclerosis, and fatty liver disease, its potential role in the pathogenesis of PPHN remains largely unexplored, highlighting a critical gap in current research.
The excessive activation of the NLR family pyrin ___domain containing 3 (NLRP3) inflammasome under hypoxic stress is a critical contributor to mitochondrial redox imbalance14. The NLRP3 inflammasome is a multiprotein complex composed of three core components: NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and the precursor of cysteinyl aspartate-specific proteinase-1 (caspase-1)15. Upon activation, the NLRP3 inflammasome triggers pyroptosis and initiates a robust inflammatory response16, leading to the maturation and release of pro-inflammatory cytokines such as interleukin (IL)-1β and IL-18 and the induction of various forms of programmed cell death17. Defects in autophagy, such as inhibition of TFEB expression, can lead to overactivation of NLRP3 inflammasomes, defective exocytosis, accumulation of cytotoxic protein aggregates, and impaired lipid degradation, thereby driving arterial endothelial injury18,19. The precise mechanisms underlying TFEB and NLRP3 inflammasome activation remain incompletely understood.
Our prior investigations demonstrated that TFEB-dependent upregulation of lysosomal biogenesis and superoxide dismutase 2 (SOD2) attenuates ROS accumulation, thereby suppressing endothelial proinflammatory activation. This study employs an integrated in vivo (late-gestation hypoxia-induced fetal rat PPHN) and in vitro (hypoxic endothelial culture) model system to dissect the TFEB-mitophagy-NLRP3 inflammasome axis. We hypothesize that TFEB regulates mitophagy and suppresses NLRP3 inflammasome activation, thereby mitigating hypoxia-induced pulmonary hypertension. Consequently, therapeutic targeting of the TFEB-NLRP3 regulatory nexus may constitute a viable strategy for PPHN management. By elucidating the role of TFEB in regulating mitochondrial quality control and inflammasome signaling, these findings advance mechanistic understanding of pulmonary vascular maladaptation while providing a molecular rationale for precision therapeutic development.
Materials and methods
Animal models of PPHN
Fifteen adult Sprague–Dawley (SD) rats (10 females, 5 males; aged 8–12 weeks) were procured from Speifer Marine Biotechnology (Beijing, China; license number: SCXK 2019-0010). The animals were maintained in a specific pathogen-free (SPF) facility at Guangdong Medical University under controlled conditions: temperature 20–25 ℃, relative humidity 50–65%, and a 12-h light/dark cycle. Standard rodent chow and filtered water were provided ad libitum, with water replenished twice weekly. For mating, females and males were housed at a 2:1 ratio.
Female rats with confirmed presence of spermatozoa (by vaginal smear) were isolated and designated as gestational day 0 (Day 0). On gestational day 19 (Day 19), pregnant rats were randomly divided into three groups: (1) Control group (normoxic conditions), (2) Hypoxia group (hypoxic conditions without intervention), and (3) Eltrombopag (EO) group (hypoxic conditions with daily intraperitoneal Eltrombopag administration at 20 mg/kg)20. Hypoxic conditions were maintained at 12% oxygen21,22 in a controlled chamber from Day 19 through Day 2123,24.
On Day 22, pregnant rats were anesthetized via isoflurane inhalation (3–4% in oxygen) until loss of consciousness, confirmed by the absence of pedal reflex. Cesarean sections were performed to deliver fetal rats, with an average litter size of 15 and fetal weight of 5 g. Fetal rats were euthanized by decapitation, and lung and heart tissues were collected for analysis. All procedures adhered to the ethical guidelines of the Animal Ethics Committee of Guangdong Medical University (Approval No: GDY2202208) in accordance with the ARRIVE guidelines. Institutional and regulatory standards for animal research were strictly followed.
Histopathological analysis and morphometric evaluation
Fresh tissues were fixed in 4% paraformaldehyde at 4 °C for 48 h. Tissues were dehydrated through a graded ethanol series, cleared in xylene, and embedded in paraffin using an automated tissue processor (Leica TP1020, Germany). Serial 4 μm sections were cut with a rotary microtome (Leica RM2235, Germany). Sections were stained with hematoxylin and eosin (H&E) as per standard protocols and examined under a light microscope (Nikon Eclipse Ci, Japan) at 40 × and 400 × magnification. Ten random fields per section were analyzed for histopathological evaluation.
Pulmonary arteriole morphometry was performed using Image-Pro Plus software (version 6.0; Media Cybernetics, USA). Key parameters included external diameter (ED), medial thickness (MT), medial area (MA), and total vascular area (TA). Vascular remodeling indices were calculated as:
-
1.
Medial area percentage: MA% = (MA/TA) × 100
-
2.
Medial thickness percentage: MT% = (2 × MT/ED) × 100
Analyses were conducted by two blinded investigators, with interobserver variability < 5% (ICC = 0.96, P < 0.001). A minimum of 20 arterioles (50–150 μm ED) per animal were evaluated.
Transmission electron microscopy (TEM)
For ultrastructural analysis, tissue samples (approximately 1 mm3) were fixed in 2.5% glutaraldehyde (0.1 M phosphate buffer, pH 7.4) at 4 °C for 24 h. Tissues were then dehydrated through a graded ethanol series, rinsed in pure acetone, and embedded in epoxy resin, followed by polymerization at 60 °C for 48 h. Ultrathin Sects. (70 nm) were cut using an ultramicrotome (Leica EM UC7, Germany), mounted on 200-mesh copper grids, and sequentially stained with 3% uranyl acetate (15 min) and Reynolds lead citrate (5 min). Ultrastructural observations were conducted using a JEM-1400Plus transmission electron microscope (JEOL Ltd., Japan) at 80 kV. Digital images were acquired with a CCD camera system (AMT XR81, USA) and analyzed for subcellular features, including mitochondrial morphology, autophagosome formation, and organelle alterations. A minimum of 20 fields per sample were examined at magnifications of 2,500 × to 10,000 × .
Immunohistochemistry
Tissue sections were dried at 60 °C, deparaffinized, and rehydrated. Antigen retrieval was conducted using heat-induced epitope retrieval in ethylenediaminetetraacetic acid (EDTA) buffer (pH 8.0), followed by endogenous peroxidase blockade with 3% H₂O₂. Sections were blocked with 10% normal goat serum (Servicebio, China) or 3% bovine serum albumin (BSA; Servicebio, China), and incubated overnight at 4 °C with primary antibodies: anti-TFEB (1:1000, Affinity, USA), anti-microtubule-associated protein 1 light chain 3 (LC3, 1:1000 dilution, Cell Signaling Technology, USA), anti-NLRP3 (1:1000 dilution, Affinity, USA), and anti-caspase-1 (1:1000 dilution, Affinity, USA), with β-actin (1:5000 dilution, Cell Signaling Technology, USA) serving as the negative control. Subsequently, sections were incubated with HRP-conjugated goat anti-rabbit secondary antibody (1:5000 dilution, Affinity, USA) for 1 h at room temperature. Detection was performed using 3,3'-diaminobenzidine (DAB), followed by hematoxylin counterstaining and mounting. Digital images were captured using a high-resolution slide scanner (Pannoramic 250 Flash III, 3DHistech) and analyzed with Image-Pro Plus 6.0 software, with a minimum of five random fields evaluated per section.
Cell culture and transfection
-
1.
Human pulmonary artery endothelial cells (HPAECs, Genio Biotechnology, China, Catalog #3100) at passage 1 were cultured in endothelial cell medium (ECM; Sciencell, USA) supplemented with 10% fetal bovine serum (FBS), 1% endothelial cell growth supplement, and 1% penicillin/streptomycin. Cells at passage 4–6 in the logarithmic growth phase were used for experiments. The study included three experimental groups:
-
2.
Control group: HPAECs cultured under normoxic conditions (37 °C, 5% CO₂, 95% air) for 24 h;
-
3.
Hypoxia group: HPAECs exposed to hypoxia (37 °C, 5% CO₂, 94% N₂, 1% O₂) for 24 h using a tri-gas incubator (Galaxy 48R, Eppendorf, Germany);
-
4.
Hypoxia + siRNA group: HPAECs transfected with gene-specific siRNAs targeting either TFEB, PINK1, or Parkin, along with a negative control siRNA (Thermo Fisher Scientific, USA), using Lipofectamine™ RNAiMAX transfection reagent (Thermo Fisher Scientific, USA). Post-transfection, cells were incubated under normoxia for 24 h, followed by hypoxia for an additional 24 h before harvesting for analysis.
Flow cytometry
Cells were collected for mitochondrial membrane potential (ΔΨm) evaluation using a commercial kit (Boxbio, Beijing, China). The fluorescent probe JC-1 was utilized following the manufacturer’s instructions. Cells were incubated with JC-1 working solution at 37 °C for 20 min in the dark. After incubation, cells were centrifuged at 300 × g for 5 min and washed twice with 1 × JC-1 assay buffer. The cell pellet was resuspended in fresh 1 × JC-1 assay buffer for analysis. ΔΨm was quantified using a BD FACSCalibur flow cytometer (Becton, Dickinson and Company, USA), with fluorescence emission measured: green fluorescence (monomeric JC-1, 530 nm) and red fluorescence (J-aggregates, 590 nm) upon excitation at 488 nm.
Immunofluorescence staining
-
1.
Tissue Section Preparation
The tissue sections were deparaffinized by sequential immersion in xylene (Servicebio, China) for 30 min, followed by rehydration through a descending ethanol gradient and rinsed in distilled water. Subsequent PBS (pH 7.4) washes were performed thrice (5 min/wash) using a laboratory shaker. After brief air-drying, specimens were circumscribed with a hydrophobic barrier pen and blocked with 3% BSA in PBS for 30 min. Sections were incubated overnight at 4 °C in a humidity-controlled chamber with primary antibody diluted in blocking buffer: anti-NLRP3, anti-caspase-1, and anti-gasdermin D (GSDMD) (all 1:100 dilution, Affinity, USA). Following PBS washes (3 × 5 min), CY3-conjugated secondary antibodies (Servicebio, China) were applied for 50 min in the dark at room temperature. After PBS washing, nuclear counterstaining was achieved through 10-min DAPI exposure in dark conditions. Sections underwent additional PBS washes and subsequent treatment with autofluorescence quencher B (5 min) followed by 10-min aqueous immersion. Sections were finally mounted with anti-fade mounting medium (Servicebio, China) and coverslipped.
-
2.
Cell Culture and Processing
Cells were seeded onto sterile glass coverslips placed in culture plates. At 60–70% confluency, hypoxia induction was initiated. Post-treatment, cells on coverslips were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were permeabilized with 0.1% Triton X-100 (Solarbio, China) for 10 min at room temperature and then blocked for 30 min. Cells were incubated overnight at 4 °C with the primary antibodies: anti-TFEB (1:100 dilution, Affinity, USA). After primary antibody incubation, the cells were treated with a fluorescence-conjugated secondary antibody, Rabbit Anti-Goat IgG (H&L)-AF647 (1:200 dilution; SAB, USA), for 1 h at room temperature in the dark. After PBS washes (3 × 5 min), nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Solarbio, China) for 5 min at room temperature in the dark. Coverslips were washed in PBS (3 × 5 min), mounted onto glass slides using an anti-fluorescence mounting medium (Solarbio, China), and visualized under an FV3000 confocal microscope (Olympus, Japan). Fluorescence imaging was conducted using the following excitation/emission parameters: 358 nm excitation and 460 nm emission for DAPI, 510 nm excitation and 590 nm emission for CY3, and 650 nm excitation and 668 nm emission for AF647. For each coverslip, three randomly selected fields of view were captured at 20 × magnification to ensure representative sampling and imaging consistency.
Western blot (WB) analysis
The samples were homogenized in 1 mL of RIPA lysis buffer (Solarbio, China) supplemented with 1% phenylmethylsulfonyl fluoride (PMSF) using a cryogenic grinder (Thermo Fisher Scientific, USA) to ensure complete protein extraction. The homogenate was centrifuged at 12,000 rpm for 20 min at 4 °C to separate the supernatant, and the total protein concentration was quantified using a bicinchoninic acid (BCA) protein assay kit (Solarbio, China). Equal amounts of protein (30 μg of total protein per lane) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 12% resolving gels, and subsequently electrotransferred onto a polyvinylidene fluoride (PVDF) membrane (Beyotime, China). The membrane was blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 2 h at room temperature to prevent non-specific binding. Following blocking, the membrane was incubated overnight at 4 °C with following specific primary antibodies (all 1:1000 dilution, Affinity, USA; unless otherwise specified): anti-TFEB, anti-PINK1, anti-Parkin, anti-NLRP3, anti-GSDMD, anti-eNOS (SAB, USA), anti-LC3 (Cell Signaling Technology, USA), anti-ASC (Abcam, UK), and anti-TOMM20 (Abcam, UK), and β-actin (Signalway Antibody, USA) or GAPDH (Signalway Antibody, USA) as loading controls. After primary antibody incubation, the membrane was washed and incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000 dilution; Affinity, USA) for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence (ECL) substrate and imaged using a Tanon-5200 Multi gel imaging system (Tianneng Company, China). Densitometric analysis was performed to quantify protein expression levels relative to β-actin or GAPDH.
qRT-PCR
RNA from treated cell samples was extracted using the Trizol method. mRNA expression levels of TFEB, PINK1 and Parkin were detected by the two-step SYBR Green-based qRT-PCR method. Vazyme HiScript® III RT SuperMix (Vazyme Biotechnology, China) was used as the reverse transcription reagent, and RT-PCR was performed on a Light Cycler 480 real-time PCR instrument (Roche, Switzerland). The qPCR protocol consisted of initial denaturation at 95 °C for 30 s (sec), 40 cycles of denaturation at 95 °C for 10 s and annealing/extension at 60 °C for 30 s, and a final melt curve stage (95 °C for 15 s, 60 °C for 60 s, 95 °C for 15 s). The 2-ΔΔCt method was used for relative quantification, with GAPDH gene as a reference. The primer sequences are shown in Table 1.
Statistical analysis
GraphPad Prism 9.0 software was used to analyze the data. Student’s t-test was used for comparisons between two groups. One-way ANOVA with Tukey’s honestly significant difference (HSD) post hoc test was used for multi-group comparisons.. Significance levels were denoted as follows: ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; ns (not significant), P > 0.05.
Results
Hypoxia upregulates TFEB expression in pulmonary arterioles of fetal rats
A rat model of PPHN was successfully established following the experimental protocol outlined in Fig. 1A. Histopathological analysis revealed characteristic changes, including inflammatory cell infiltration, interstitial tissue hyperplasia, vascular remodeling, and thickening of the vascular smooth muscle layer (Fig. 1B). Quantitative analysis demonstrated that, compared to the control group, the hypoxia group exhibited statistically significant increases in MA% and MT% of pulmonary arterioles (Fig. 1C). Furthermore, the hypoxia group displayed pathological features such as right ventricular wall thickening, disorganized myocardial cell arrangement, and an increased number of apoptotic cells (Fig. 1D,E). Immunohistochemistry revealed a significantly higher proportion of TFEB-positive cells in pulmonary arteriolar endothelial cells, alveolar epithelial cells, and bronchial smooth muscle in the hypoxia group compared to the control group, with significantly elevated relative optical density values (Fig. 1F,G). WB analysis further confirmed significant upregulation of TFEB protein expression in hypoxic pulmonary tissues (Fig. 1H,I, and Supplementary Table 1). These findings suggest that TFEB may play a critical regulatory role in hypoxia-induced pulmonary vascular injury.

Hypoxia induces upregulation of TFEB expression in pulmonary arterioles of fetal rats (A) Schematic representation of the PPHN modeling procedure. EO: Eltrombopag. (B) Representative HE-stained lung tissue sections from control and hypoxia groups, illustrating pathological alterations in pulmonary microvessels. Scale bars: 500 µm and 50 µm. Black arrows indicate pulmonary arterioles; Yellow arrows indicate neutrophils; Red arrows indicate bronchial smooth muscle. (C) Quantitative comparison of MA% and MT% between the two groups. MA: medial area, MT: medial thickness (n = 5 per group). (D) Representative HE-stained longitudinal sections of the whole heart from the two groups, demonstrating myocardial pathological changes. Scale bar: 50 µm. Black arrows indicate necrotic cardiomyocytes. (E) Comparison of right ventricular wall thickness between the two groups (n = 5). (F) Representative immunohistochemical staining of TFEB in lung tissues between the two groups. Scale bar: 50 µm. (G) Quantitative comparison of the average optical density of TFEB staining between the two groups. (H) Western blot analysis of TFEB protein expression between the two groups (n = 3). (I) Comparison of relative TFEB protein expression levels between the two groups (n = 3). ** P < 0.01; **** P < 0.0001.
Hypoxia activates mitophagy in pulmonary arterioles of fetal rats
To investigate the role of mitophagy in hypoxia-mediated injury, we analyzed ultrastructural changes via TEM. In the control group, endothelial cells exhibited typical morphological characteristics, including intact organelles, well-defined nuclear membranes, and uniformly distributed chromatin. In contrast, endothelial cells in the hypoxia group displayed significant ultrastructural damage, characterized by abnormal nuclear morphology, severely swollen mitochondria with structural disorganization, and the presence of numerous mitophagosomes and autolysosomes (Fig. 2A). Quantification showed reduced mitochondrial numbers, but significantly increased autophagosomes/autolysosomes in the hypoxia group (Fig. 2B).

Hypoxia activates mitophagy in pulmonary arterioles of fetal rats (A) Representative transmission electron microscopy images of pulmonary arteriole endothelial cells from control and hypoxia groups, showing mitochondrial morphology. Scale bars: 5 µm and 1 µm. Yellow arrows indicate the nucleus and nuclear membrane; Red arrows indicate mitochondria; Blue arrows indicate autophagosomes and autolysosomes. (B) Quantitative comparison of mitochondria and autophagosomes/lysosomes between the two groups (n = 5 per group). (C) Representative immunohistochemical staining of LC3 in lung tissues between the two groups. Scale bar: 50 µm. (D) Quantitative comparison of the average optical density of LC3 staining between the two groups (n = 5 per group). (E) Western blot analysis of mitophagy-related proteins in lung tissues between the two groups (n = 3). (F) Comparison of relative protein expression levels of mitophagy-related proteins between the two groups (n = 3). * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Immunohistochemistry revealed a significantly higher density of LC3-positive perivascular cells (Fig. 2C) and increased mean optical density (Fig. 2D) in the hypoxia group. WB analysis showed the upregulated expression of mitophagy-related proteins, including PINK1, Parkin, TOMM20, COX IV, P62, and LC3II/I ratio in thehypoxia group (Fig. 2E,F, and Supplementary Table 2). Collectively, these findings provide robust morphological and molecular evidence that hypoxia significantly enhances mitophagy activity in the pulmonary arterioles of fetal rats.
Hypoxia activates the NLRP3 inflammasome in pulmonary arterioles of fetal rats
To further investigate hypoxia-driven the mechanisms in pulmonary arterioles, we investigated whether hypoxia exacerbates pulmonary vascular injury through the activation of the NLRP3 inflammasome. Immunohistochemistry demonstrated a significantly increased proportion of NLRP3-positive cells in the pulmonary tissues of the hypoxia group, with positive signals predominantly localized around pulmonary arterioles (Fig. 3A). The relative optical density values were also significantly higher in the hypoxia group (Fig. 3B). Similarly, the proportion of caspase-1-positive cells was significantly increased, with elevated optical density in the hypoxia group (Fig. 3C,D). WB analysis confirmed significant upregulation of core NLRP3 inflammasome components (NLRP3, caspase-1, ASC, IL-1β, and GSDMD) in hypoxic pulmonary tissues, accompanied by a concomitant reduction in eNOS expression (Fig. 3E,F, and Supplementary Table 3). These results demonstrate that hypoxia plays a pivotal role in pulmonary vascular injury by activating the NLRP3 inflammasome pathway.

Hypoxia activates the NLRP3 inflammasome in pulmonary arterioles of fetal rats (A) Representative immunohistochemical staining of NLRP3 in lung tissues from control and hypoxia groups. Scale bar: 50 µm. Black arrows indicate pulmonary arterioles; Red arrows indicate bronchi. (B) Quantitative comparison of the average optical density of NLRP3 staining between the two groups (n = 5 per group). (C) Representative immunohistochemical staining of caspase-1 in lung tissues from the two groups. Scale bar: 50 µm. Black arrows indicate positive stained cells. (D) Quantitative comparison of the average optical density of caspase-1 staining between the two groups (n = 5 per group). (E) Western blot analysis of NLRP3 inflammasome-related proteins in lung tissues from the two groups. (F) Comparison of relative protein expression levels of NLRP3 inflammasome-related proteins between the two groups (n = 3). * P < 0.05; ** P < 0.01; *** P < 0.001.
Inhibition of TFEB expression suppresses mitophagy and activates the NLRP3 inflammasome
To further clarify the role of TFEB in hypoxia-induced pulmonary vascular endothelial cell injury, we investigated the regulatory mechanisms by which TFEB expression modulates mitophagy and NLRP3 inflammasome activation. Prolonged hypoxia induced global TFEB upregulation in HPAECs, with predominant cytosolic accumulation (Fig. 4A). Quantification confirmed that TFEB levels were significantly elevated in the hypoxia group (Fig. 4B). qRT-PCR analysis revealed an upregulation in the mRNA expression levels of TFEB, PINK1, and Parkin in the hypoxia group (Fig. 4C). Flow cytometry indicated a significant reduction in ΔΨm in the hypoxia group, which was further exacerbated by TFEB knockdown (si-TFEB group) (Fig. 4D,E). Compared to the si-NC group, TFEB inhibition significantly decreased the levels of mitophagy-related proteins, including PINK1, Parkin, TOMM20, COX IV, P62, and LC3II/I ratio, indicating suppressed mitophagy activity (Fig. 4F,G, and Supplementary Table 4).

Hypoxia induces TFEB expression, and inhibition of TFEB suppresses mitophagy in HPAECs (A) Representative immunofluorescence images of single-labeling TFEB in control and hypoxia-treated HPAECs (n = 3 per group). Scale bar = 50 µm. (B) Quantitative comparison of the average optical density of TFEB staining among the groups (n = 5 per group). (C) Comparison of mRNA expression levels of TFEB, PINK1, and Parkin among the groups. HPAECs were transfected with siRNAs targeting TFEB, PINK1, or Parkin, along with a negative control siRNA (Thermo Fisher Scientific, USA), using Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, USA). After transfection, cells were incubated under normoxia for 24 h, followed by hypoxia for additional 24 h before harvesting for analysis. (D) Flow cytometry analysis of mitochondrial membrane potential (ΔΨm) in each group (n = 3 per group). (E) Quantitative comparison of mitochondrial membrane potential (ΔΨm) among the groups. (F) Western blot analysis of TFEB and mitophagy-related proteins among the groups (n = 3). (G) Comparison of relative protein expression levels of TFEB and mitophagy-related proteins among the groups (n = 3). ns: not significant; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Notably, inhibition of PINK1 or Parkin expression significantly upregulated the levels of GSDMD. TFEB knockdown also elevated NLRP3, eNOS and GSDMD expression (Fig. 5A,B, and Supplementary Table 5), suggesting enhanced NLRP3 inflammasome activation and pyroptosis. These findings demonstrate that TFEB plays a pivotal role in regulating mitophagy and NLRP3 inflammasome activation. Suppression of TFEB expression exacerbates hypoxia-induced pyroptosis in HPAECs.

Inhibition of TFEB activates the NLRP3 inflammasome in HPAECs (A) Western blot analysis of NLRP3 inflammasome-related proteins in the groups (n = 3). (B) Comparison of relative protein expression levels of NLRP3 inflammasome-related proteins among the groups (n = 3). ns: not significant; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Eltrombopag treatment suppresses mitophagy and enhances NLRP3 inflammasome activation
To explore the role of TFEB-mediated mitophagy in regulating NLRP3 inflammasome activation, we performed intervention experiments using the TFEB inhibitor Eltrombopag (EO). Histopathological analysis revealed exacerbated lung injury in the EO group versus the hypoxia group, characterized by increased inflammatory cell infiltration, aggravated interstitial tissue hyperplasia, pronounced vascular remodeling, and marked thickening of the vascular smooth muscle layer (Fig. 6A). Quantitative analysis demonstrated significantly elevated MA% and MT% in pulmonary arterioles of the EO group (Fig. 6B). TEM revealed abnormal nuclear morphology, including severe mitochondrial swelling and structural distortion, and a significant reduction in the number of mitophagosomes and autolysosomes in the EO group (Fig. 6C). Quantification confirmed an increased number of mitochondria in the EO group, with a decreased number of autophagosomes/autolysosomes (Fig. 6D).

Eltrombopag treatment suppresses mitophagy and enhances NLRP3 inflammasome activation in fetal rats (A) Representative HE-stained lung tissue sections from the groups, showing pathological changes in pulmonary microvessels. Scale bars: 500 µm and 50 µm. Black arrows indicate pulmonary arterioles; Yellow arrows indicate neutrophils; Red arrows indicate bronchial smooth muscle. (B) Quantitative comparison of MA% and MT% among the groups. MA: medial area, MT: medial thickness (n = 5 per group). (C) Representative transmission electron microscopy images of pulmonary arteriole endothelial cells from the groups, illustrating mitochondrial morphology. Scale bars: 5 µm and 1 µm. Yellow arrows indicate the nucleus and nuclear membrane; Red arrows indicate mitochondria; Blue arrows indicate autophagosomes and autolysosomes. (D) Quantitative comparison of mitochondria and autophagosomes/lysosomes among the groups (n = 5 per group).
Immunofluorescence revealed a significant increase in the number of NLRP3-, caspase-1-, and GSDMD-positive cells in the EO group (Fig. 7A,B), with markedly higher mean optical density values, compared to the Hypoxia group (Fig. 7C). WB analysis demonstrated that, the expression levels of mitophagy-related proteins, including PINK1, Parkin, TOMM20, COX IV, P62, and LC3, were significantly downregulated in the EO group, relative to the hypoxia group (Fig. 8A,B, and Supplementary Table 6). Conversely, the expression levels of NLRP3 inflammasome-related proteins, including NLRP3, caspase-1, ASC, IL-1β, and GSDMD, were significantly upregulated in the EO group (Fig. 8C,D, and Supplementary Table 7). These results indicate that inhibition of TFEB expression suppresses mitophagy activity, enhances NLRP3 inflammasome activation, and promotes pyroptosis.

Upregulated expression of NLRP3, caspase-1, and GSDMD in fetal rats following TFEB inhibition as revealed by immunofluorescence staining (A) Representative immunofluorescence images of single-labeling NLRP3 and caspase-1 from the groups (n = 3 per group). Scale bar = 50 µm. White arrows indicate pulmonary arterioles. (B) Representative immunofluorescence images of single-labeling GSDMD from the groups (n = 3 per group). Scale bar = 50 µm. White arrows indicate pulmonary arterioles. (C) Quantitative comparison of the average optical density of NLRP3, caspase-1, and GSDMD staining among the groups (n = 5 per group). ns: not significant; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.

Eltrombopag treatment reduces mitophagy-related protein expression and enhances NLRP3 inflammasome protein expression in fetal rats (A) Western blot analysis of TFEB and mitophagy-related proteins in the groups (n = 3). (B) Comparison of relative protein expression levels of TFEB and mitophagy-related proteins among the groups (n = 3). (C) Western blot analysis of NLRP3 inflammasome-related proteins in the groups (n = 3). (D) Comparison of relative protein expression levels of NLRP3 inflammasome-related proteins among the groups (n = 3). ns: not significant; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Discussion
This study provided novel insights into the molecular mechanisms underlying PPHN, demonstrating that hypoxia-induced PPHN in fetal rats was closely associated with enhanced mitophagy and NLRP3 inflammasome activation. Crucially, we revealed that inhibition of TFEB not only suppresses mitophagy but also promotes NLRP3 inflammasome activation and upregulates associated inflammatory factors, suggesting a critical role of the mitophagy-NLRP3 inflammasome signaling axis in the pathogenesis of PPHN. These findings underscore TFEB as a pivotal regulator coupling mitochondrial clearance to inflammatory responses in PPHN. We propose that TFEB activation enhances mitophagic activity, attenuates inflammatory cascades, and alleviates clinical manifestations of pulmonary hypertension, offering a novel therapeutic strategy.
In PPHN, mitochondrial dysfunction drives heightened mitophagy, exacerbating pulmonary vascular injury and dysfunction while promoting hypoxia-mediated apoptosis25,26,27,28. Hypoxia-induced reactive oxygen species (ROS) activate the PINK1/Parkin-mediated mitophagy pathway, contributing to pulmonary vascular remodeling29. Additionally, impaired crosstalk between autophagy and inflammation, exemplified by oxidized low-density lipoprotein disrupting the TFEB-P300-BRD4 axis, leads to defective macrophage autophagy, cytosolic TFEB accumulation, elevated ROS, and inflammatory gene overexpression30.
In a cardiac model, hypoxia/reoxygenation induces cardiomyocyte apoptosis31, whereas upregulation of TFEB mitigates hypoxic-ischemic injury via lysosomal autophagy pathway32. In an arteriosclerosis model, TFEB overexpression rescues lipid-induced lysosomal dysfunction and mitigated downstream pathological consequences by upregulating the expression of autophagy-lysosomal pathway-related genes33. Furthermore, TFEB has been reported to effectively attenuate mitophagy by regulating the PINK1/Parkin signaling pathway, thereby alleviating intestinal barrier damage caused by oxidative stress34. In this study, the immunofluorescence co-localization analyses revealed that hypoxic stress induces predominant cytosolic retention of TFEB. WB confirmed that elevated TFEB expression correlated with upregulated mitophagy-related proteins. Additionally, TFEB knockdown exacerbated hypoxia-induced ΔΨm dissipation and upregulated the expression of mitophagy markers (PINK1, Parkin, TOMM20, COX IV, and LC3-II/I ratio). These data demonstrate TFEB’s dependency on the PINK1/Parkin pathway to orchestrate mitochondrial quality control.
A key mechanism driving the development of PPHN is mitochondrial oxidative stress35. Restoring mitochondrial redox balance represents a critical therapeutic strategy for PPHN. Mitochondria undergo fusion via coordinated actions of inner and outer membrane proteins, forming interconnected networks that facilitate mitochondrial DNA (mtDNA) repair36. During fission, damaged mtDNA is segregated into mitochondria with low ΔΨm, where it is recognized by mitochondrial quality control proteins and selectively cleared via mitophagy37. The permeability and functional integrity of the mitochondrial membrane play a pivotal role in maintaining ΔΨm. In this study, hypoxia-induced ΔΨm reduction reflected mitochondrial dysfunction and mitophagy activation. Notably, TFEB knockdown further decreased ΔΨm in the si-TFEB group, indicating a role of TFEB in regulating mitophagic efficiency. This finding provides a novel experimental foundation for the development of targeted therapeutic interventions, highlighting TFEB as a potential regulator of mitochondrial quality control in PPHN.
The activation of the NLRP3 inflammasome significantly amplifies the release of inflammatory factors, exacerbates oxidative stress damage, and worsens pulmonary vascular endothelial injury. Therefore, inhibiting the activation of the NLRP3 inflammasome suppresses endothelial injury inflammation and oxidative damage, which strategy may be an important for treating PPHN38. Elevated expression of NLRP3 associates with mitochondrial damage and mitophagic impairment39,40. Modulation of PINK1/Parkin-dependent mitophagy has been shown to significantly reduce NLRP3 expression41,42, consistent with our findings. Notably, TFEB activity exhibits temporal dynamics under hypoxic conditions. In the cerebral ischemia models of rats, TFEB is activated during the early ischemic phase, evidenced by significant upregulation of protein and nuclear translocation (peaking at 6 h of post-occlusion)43. This activation promotes neuronal survival and protection against ischemic damage. As ischemia progresses, TFEB function gradually declines, characterized by reduced nuclear accumulation and predominant cytoplasmic retention. This functional impairment ultimately contributes to ischemic injury, particularly during later ischemia stages. Similarly, we observed predominantly cytoplasmic retention of TFEB in HPAECs after 24 h hypoxia, indicating functional impairment. This dysfunctional TFEB state contributes to pulmonary hypertension by exacerbating NLRP3-mediated pyroptosis.
Importantly, TFEB serves as a master negative regulator of the NLRP3 inflammasome. The NLRP3-driven inflammatory response has been identified as a critical factor in asthma pathogenesis, where TFEB activation restores impaired autophagy, mitigates NLRP3-mediated lung inflammation, and ameliorates asthma phenotypes44. In a Parkinson’s disease model, p38 was found to activate the NLRP3 inflammasome by suppressing TFEB in microglia, while TFEB signaling inhibits NLRP3 inflammasome via upregulation of LAMP2A45. Meanwhile, mitochondrial dysfunction primes NLRP3 activation, while TFEB-driven mitochondrial quality control via PINK1/Parkin signaling restores homeostasis46. In this study, hypoxia significantly upregulated NLRP3, caspase-1, and ASC expression. TFEB inhibition markedly reduced mitophagy markers (e.g., PINK1, Parkin, LC3-II/I ratio), while concomitantly amplifying NLRP3 inflammasome components and pyroptosis. This established TFEB-mediated mitophagy as a regulatory mechanism for NLRP3 suppression.TFEB is a pivotal anti-inflammatory effector whose therapeutic activation may counteract NLRP3-driven inflammatory pathology through enhanced mitochondrial clearance, positioning it as a promising therapeutic target.
It is important to acknowledge several limitations in the current study, including the lack of genetic knockout rat models. Future investigations utilizing modulators of mitophagy and inflammasome would provide further mechanistic insights. Although we demonstrate that TFEB modulates the mitophagy-NLRP3 inflammasome axis via the PINK1/Parkin pathway in hypoxia-induced PPHN, further research is necessary, including investigating whether TFEB interacts with other autophagy pathways under hypoxia, and potential crosstalk with alternative oxidative stress and inflammatory signaling pathways.
Conclusion
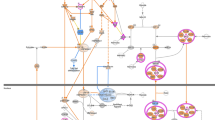
In summary, this study demonstrates that TFEB regulates PINK1/Parkin-mediated mitophagy in PPHN. Hypoxia-induced mitochondrial damage activates the NLRP3 inflammasome. Inhibition of TFEB disrupts the mitophagy-mediated clearance of damaged mitochondria, leading to enhanced activation of NLRP3 inflammasome and subsequent exacerbated tissue injury (Fig. 9). These findings establish TFEB as a pivotal regulator of the mitophagy-NLRP3 inflammasome axis, highlighting its protective role in PPHN pathogenesis.

Mechanism of TFEB in Suppressing NLRP3 Inflammasome Activation via Mitophagy. Hypoxia induces inflammation and pyroptosis through mitochondrial damage, which activates the NLRP3 inflammasome, and promotes the secretion of IL-1β and GSDMD. TFEB inhibits NLRP3 inflammasome activation by enhancing mitophagy, which clears damaged mitochondria. Conversely, TFEB inhibition impairs mitophagy, leading to accumulation of damaged mitochondria, thereby exacerbating NLRP3 inflammasome activation and tissue injury.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Singh, Y. & Lakshminrusimha, S. Pathophysiology and management of persistent pulmonary hypertension of the newborn. Clin. Perinatol. 48, 595–618. https://doi.org/10.1016/j.clp.2021.05.009 (2021).
Farrow, K. N. et al. Mitochondrial oxidant stress increases PDE5 activity in persistent pulmonary hypertension of the newborn. Respir. Physiol. Neurobiol. 174, 272–281. https://doi.org/10.1016/j.resp.2010.08.018 (2010).
Culley, M. K. & Chan, S. Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 128, 3704–3715. https://doi.org/10.1172/jci120847 (2018).
He, J. et al. Inhibition of mitochondrial oxidative damage improves reendothelialization capacity of endothelial progenitor cells via SIRT3 (Sirtuin 3)-enhanced SOD2 (superoxide dismutase 2) deacetylation in hypertension. Arterioscler. Thromb. Vasc. Biol. 39, 1682–1698. https://doi.org/10.1161/atvbaha.119.312613 (2019).
Linqing, L. et al. Hypoxia-induced PINK1/Parkin-mediated mitophagy promotes pulmonary vascular remodeling. Biochem. Biophys. Res. Commun. 534, 568–575. https://doi.org/10.1016/j.bbrc.2020.11.040 (2021).
Chen, M. et al. TFEB biology and agonists at a glance. Cells https://doi.org/10.3390/cells10020333 (2021).
Cora, D., Bussolino, F. & Doronzo, G. TFEB signalling-related MICRORNAS and autophagy. Biomolecules https://doi.org/10.3390/biom11070985 (2021).
Huang, M. et al. 3-Hydroxybutyrate ameliorates sepsis-associated acute lung injury by promoting autophagy through the activation of GPR109α in macrophages. Biochem. Pharmacol. 213, 115632. https://doi.org/10.1016/j.bcp.2023.115632 (2023).
Dombi, E., Mortiboys, H. & Poulton, J. Modulating mitophagy in mitochondrial disease. Curr. Med. Chem. 25, 5597–5612. https://doi.org/10.2174/0929867324666170616101741 (2018).
Kane, L. A. et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell. Biol. 205, 143–153. https://doi.org/10.1083/jcb.201402104 (2014).
Ma, C. et al. Ubiquitinated AIF is a major mediator of hypoxia-induced mitochondrial dysfunction and pulmonary artery smooth muscle cell proliferation. Cell Biosci. 12, 9. https://doi.org/10.1186/s13578-022-00744-3 (2022).
Liu, W., Li, Y., Bo, L., Li, C. & Jin, F. Positive regulation of TFEB and mitophagy by PGC-1α to alleviate LPS-induced acute lung injury in rats. Biochem. Biophys. Res. Commun. 577, 1–5. https://doi.org/10.1016/j.bbrc.2021.08.064 (2021).
Park, K. et al. Impaired TFEB activation and mitophagy as a cause of PPP3/calcineurin inhibitor-induced pancreatic β-cell dysfunction. Autophagy 19, 1444–1458. https://doi.org/10.1080/15548627.2022.2132686 (2023).
Mavrogiannis, E. et al. Pirfenidone ameliorates pulmonary arterial pressure and neointimal remodeling in experimental pulmonary arterial hypertension by suppressing NLRP3 inflammasome activation. Pulm Circ 12, e12101. https://doi.org/10.1002/pul2.12101 (2022).
Kelley, N., Jeltema, D., Duan, Y. & He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20133328 (2019).
Takahashi, M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc. Res. 118, 372–385. https://doi.org/10.1093/cvr/cvab010 (2022).
Zhu, Y. et al. The interplay between pattern recognition receptors and autophagy in inflammation. Adv. Exp. Med. Biol. 1209, 79–108. https://doi.org/10.1007/978-981-15-0606-2_6 (2019).
Ahmed, S., Kwatra, M., Ranjan Panda, S., Murty, U. S. N. & Naidu, V. G. M. Andrographolide suppresses NLRP3 inflammasome activation in microglia through induction of parkin-mediated mitophagy in in-vitro and in-vivo models of Parkinson disease. Brain Behav. Immun. 91, 142–158. https://doi.org/10.1016/j.bbi.2020.09.017 (2021).
Evans, T. D., Jeong, S. J., Zhang, X., Sergin, I. & Razani, B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 14, 724–726. https://doi.org/10.1080/15548627.2018.1434373 (2018).
Lin, Y. et al. A small-molecule drug inhibits autophagy gene expression through the central regulator TFEB. Proc. Natl. Acad. Sci. USA 120, e2213670120. https://doi.org/10.1073/pnas.2213670120 (2023).
Deruelle, P. et al. BAY 41–2272, a direct activator of soluble guanylate cyclase, reduces right ventricular hypertrophy and prevents pulmonary vascular remodeling during chronic hypoxia in neonatal rats. Biol. Neonate 90, 135–144. https://doi.org/10.1159/000092518 (2006).
Xu, X. F. et al. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 28, 2227–2235. https://doi.org/10.1097/HJH.0b013e32833e08f1 (2010).
Xu, X. F., Gu, W. Z., Wu, X. L., Li, R. Y. & Du, L. Z. Fetal pulmonary vascular remodeling in a rat model induced by hypoxia and indomethacin. J. Matern. Fetal Neonatal Med. 24, 172–182. https://doi.org/10.3109/14767058.2010.482608 (2011).
Du, Y. et al. Altered expression of PPAR-γ and TRPC in neonatal rats with persistent pulmonary hypertension. Mol. Med. Rep. 16, 1117–1124. https://doi.org/10.3892/mmr.2017.6744 (2017).
Sharma, A., Ahmad, S., Ahmad, T., Ali, S. & Syed, M. A. Mitochondrial dynamics and mitophagy in lung disorders. Life Sci. 284, 119876. https://doi.org/10.1016/j.lfs.2021.119876 (2021).
Zhang, W. et al. Mitochondrial dysfunction in pulmonary arterial hypertension. Front. Physiol. 13, 1079989. https://doi.org/10.3389/fphys.2022.1079989 (2022).
Ornatowski, W. et al. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 36, 101679. https://doi.org/10.1016/j.redox.2020.101679 (2020).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108. https://doi.org/10.1038/emboj.2012.32 (2012).
Chen, C. et al. Effect and mechanism of PINK1/parkin-mediated mitochondrial autophagy in rat lung injury induced by nano lanthanum oxide. Nanomaterials (Basel) https://doi.org/10.3390/nano12152594 (2022).
Li, X. et al. Autophagy enhanced by curcumin ameliorates inflammation in atherogenesis via the TFEB-P300-BRD4 axis. Acta Pharm. Sin. B 12, 2280–2299. https://doi.org/10.1016/j.apsb.2021.12.014 (2022).
Song, H. et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15, 1419–1437. https://doi.org/10.1080/15548627.2019.1586246 (2019).
Chen, D. et al. Elabela ameliorates doxorubicin-induced cardiotoxicity by promoting autophagic flux through TFEB pathway. Pharmacol. Res. 178, 106186. https://doi.org/10.1016/j.phrs.2022.106186 (2022).
Fang, S. et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. 12, 88. https://doi.org/10.1038/s41419-020-03357-1 (2021).
Cao, S. et al. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK-TFEB signal pathway. Free Radic. Biol. Med. 147, 8–22. https://doi.org/10.1016/j.freeradbiomed.2019.12.004 (2020).
Tan, S. et al. Pomegranate activates TFEB to promote autophagy-lysosomal fitness and mitophagy. Sci. Rep. 9, 727. https://doi.org/10.1038/s41598-018-37400-1 (2019).
Archer, S. L. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251. https://doi.org/10.1056/NEJMra1215233 (2013).
Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. https://doi.org/10.1038/nrm3028 (2011).
Gupta, S., Cassel, S. L., Sutterwala, F. S. & Dagvadorj, J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol. Rev. 329, e13410. https://doi.org/10.1111/imr.13410 (2025).
Harris, J., Deen, N., Zamani, S. & Hasnat, M. A. Mitophagy and the release of inflammatory cytokines. Mitochondrion 41, 2–8. https://doi.org/10.1016/j.mito.2017.10.009 (2018).
Lin, Q. et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 17, 2975–2990. https://doi.org/10.1080/15548627.2020.1848971 (2021).
Wu, X. et al. NLRP3 deficiency protects against intermittent hypoxia-induced neuroinflammation and mitochondrial ROS by promoting the PINK1-parkin pathway of mitophagy in a murine model of sleep apnea. Front. Immunol. 12, 628168. https://doi.org/10.3389/fimmu.2021.628168 (2021).
Fan, L. et al. Melatonin ameliorates the progression of alzheimer’s disease by inducing TFEB nuclear translocation, promoting mitophagy, and regulating NLRP3 inflammasome activity. Biomed. Res. Int. 2022, 8099459. https://doi.org/10.1155/2022/8099459 (2022).
Liu, Y. et al. Neuronal-targeted TFEB rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy 15, 493–509. https://doi.org/10.1080/15548627.2018.1531196 (2019).
Theofani, E. et al. TFEB signaling attenuates NLRP3-driven inflammatory responses in severe asthma. Allergy 77, 2131–2146. https://doi.org/10.1111/all.15221 (2022).
Lin, W. et al. TNEA therapy promotes the autophagic degradation of NLRP3 inflammasome in a transgenic mouse model of Alzheimer’s disease via TFEB/TFE3 activation. J. Neuroinflam. 20, 21. https://doi.org/10.1186/s12974-023-02698-w (2023).
Tan, A., Prasad, R., Lee, C. & Jho, E. H. Past, present, and future perspectives of transcription factor EB (TFEB): Mechanisms of regulation and association with disease. Cell Death Differ. 29, 1433–1449. https://doi.org/10.1038/s41418-022-01028-6 (2022).
Funding
This work was supported by the science and technology project of Zhanjiang City (2022A01181); Collaborative Fund of Guangdong Basic and Applied Basic Research (2019A1515110564); Guangdong Medical Research Foundation (A2020286); the high-level talents scientific research start-up funds of the Affiliated Hospital of Guangdong Medical University (GCC2024024); Affiliated Hospital of Guangdong Medical University “Clinical Medicine plus” Scientific Research Cooperation Project; Affiliated Hospital of Guangdong Medical University Clinical Research Program (LCYJ2020DL01); Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-communicable Diseases (2022B1212030003); Open funding of Affiliated Hospital of Guangdong Medical University-research of autophagy and diseases.
Author information
Authors and Affiliations
Contributions
Ao Dang and Li Chengyan designed the experiments. Chen Chaohong, Xie Zaoye, Chen Yinhui, and Liu Ling performed the experiments. Chen Chaohong and Li Chengyan analyzed the data and prepared the manuscript. Li Chengyan edited and revised the manuscript. All authors approved the version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal experiments met the standards of the Animal Ethics Committee of Guangdong Medical University and were reviewed and approved by the Animal Ethics Committee of Guangdong Medical University (Approval Number: GDY2202208).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, C., Xie, Z., Ao, D. et al. Transcription factor EB improves hypoxic pulmonary hypertension in fetal rats by suppressing NLRP3 inflammasome activation via induction of mitophagy. Sci Rep 15, 20356 (2025). https://doi.org/10.1038/s41598-025-07068-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-07068-5
