
Abstract
The blood vessel network is heavily impacted by SARS-CoV-2 infection. How SARS-CoV-2 contributes to vascular inflammation and whether epithelio-endothelial cross-talk is involved remain unclear. We investigated in detail the interaction between SARS-CoV-2 and the vasculature using 2D and 3D vesseloid in vitro models. We first assessed whether SARS-CoV-2 is able to directly infect endothelial cells. In the absence of ACE2 in endothelial cells, no productive infection was detected. Low uptake of viral particles by ACE2-overexpressing endothelial cells was observed without efficient viral production. Thus, the indirect effect of SARS-CoV-2 infection may involve epithelio-endothelial cell cross-talk. After infection of the epithelial cells, a significant inflammatory response was detected in the endothelial cells. CXCL10 was the most highly expressed proinflammatory cytokine involved in this intercellular communication, and its function was subsequently explored. Finally, the clinical relevance of our findings was confirmed in two patient cohorts.
Similar content being viewed by others


Introduction
The coronavirus disease 2019 (COVID-19) pandemic erupted in December 2019 and rapidly evolved into one of the most complex and disruptive public health crises of the twenty-first century. The virus responsible for severe acute respiratory syndrome spread globally, triggering a pandemic in 2020. This situation has exposed vulnerabilities in the global healthcare system and disrupted economies, endangering the lives of millions of people1.
Postmortem biopsy studies first focused on lung tissue and revealed endothelial injuries with cytoplasmic vacuolization, cell detachment in pulmonary arteries and swelling2,3, accompanied with an accumulation of infiltrating immune cells, such as macrophages and lymphocytes4.
Analysis of SARS-CoV-2 infected patient serum samples revealed an increased production of proinflammatory cytokines, a phenomenon called “cytokine storm”, associated with high circulatory levels of shed adhesion molecules, such as VCAM-1 or ICAM-15. This cytokine storm may be attributed to a deregulated immune response leading to acute respiratory distress syndrome6. This syndrome is characterized by a severe inflammatory response, resulting in blood vessel inflammation, impairment of the alveolar-capillary barrier, increased vascular permeability, and disruption of gas exchange, leading to edema and death7. Furthermore, vascular thrombotic events are also well documented during SARS-CoV-2 infection, reflecting endothelial cell activation and leading to vascular injury. However, the specific molecular mechanisms involved are not fully understood.
In general, SARS-CoV-2 entry into cells requires the ACE2 receptor. The ACE2 receptor was initially identified for its role in regulating blood pressure and cardiovascular homeostasis but has become a major research focus due to its essential role as a virus entry point. Studies have shown that ACE2 is widely distributed in various tissues and is found mainly in the heart, lung, kidneys, and gastrointestinal tract, which could explain the diversity of symptoms encountered in SARS-CoV-2 infected patients8,9. However, studies have reported contradictory findings regarding the presence of ACE2 receptors in endothelial cells9,10. Lately, some reports suggested an ACE2 independent SARS-CoV-2 infection mechanism, which might explain minimal epithelial and possibly endothelial infection11. In addition, some studies have described the presence of SARS-CoV-2 RNA in coronary artery plaques or lung endothelial cell associated macrophages12,13,14, while other studies have reported endothelial cell injury without the presence of SARS-CoV-2 particles in postmortem samples from SARS-CoV-2 infected patients3.
Therefore, it is imperative to understand the mechanisms governing the effects of this virus on the vascular system. This understanding is crucial for the development of long-term treatment strategies for SARS-CoV-2 infected patients and to anticipate the evolution of new variants.
In our study, we used various experimental settings, including 2D and 3D endothelial cell models as well as cocultures with lung epithelial cell line and/or myeloid cells. We also compared our findings to the clinical situation by the dosage of patients’ blood samples and via in silico analysis of an external cohort. Taken together, our data point to an indirect mechanism of action for SARS-CoV-2 on the vascular endothelium involving an epithelial-endothelial cross-talk.
Results
Absence of direct effects of SARS-CoV-2 on the vascular endothelium
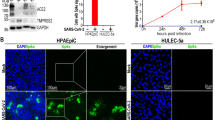
To determine the impact of the virus on the vascular endothelium, we cultured various human endothelial primary cell lines, including human umbilical vein endothelial cells (HUVECs), human umbilical artery endothelial cells (HUAECs), and human coronary artery endothelial cells (HCAECs). No ACE2 protein expression was detected in these cell types by Western blotting. In contrast, the expression levels of these genes in the epithelial human lung cancer cell line Calu-3 (Fig. 1A) were very high. To investigate whether ACE2 could serve as a functional receptor in endothelial cells for SARS-CoV-2 entry, we overexpressed ACE2 in a human umbilical vein endothelial cell line (ACE2-HUVECs) (Fig. 1A). ACE2-HUVECs were then used as the cellular component in the vesseloid model and compared to non-ACE2-overexpressing HUVECs (WT-HUVECs). The vesseloid model was described by our group in a previous publication and consists of a 3D tubular structure harboring an endothelial cell lining that mimics the configuration of a physiological vessel15. SARS-CoV-2 infection was performed, and the mRNA levels of the N protein, the nucleocapsid of the virus, in the supernatant were analyzed. In ACE2-HUVECs, viral infection was very low, and minimal viral particle production was observed after 3 or 4 days in the supernatant (Fig. 1B). In Calu-3 cells, the N protein concentration was approximately two log greater (Fig. 1B). A time-dependent analysis of the infection efficiency was further performed (with an MOI of 1 or an MOI of 0.1) on Calu-3 cells, and the peak N gene expression was detected at 3 days post infection (Sup Fig. 1A). The behavior of control HUVECs, HCAECs and HUAECs was similar to that of ACE2-HUVECs (Fig. 1B; Sup Fig. 1B).

Absence of direct effects of SARS-CoV-2 on the vascular endothelium. (A) Western blot analysis of the ACE2 protein in HUVECs, HUAECs, HCAECs, Calu-3 cells and ACE2 overexpressing HUVECs cells. Tubulin was used as a control. (B) RT‒qPCR quantification of N gene RNA in media after vesseloid or Calu-3 cell SARS-CoV-2 infection. Analysis was performed at 3- or 4-days post infection (MOI = 0.1). The values were normalized to a range of RNA extracted from the virus we use to infect, for which we know the TCID50 and PFU. The results are presented as the mean ± SEM (one-way ANOVA *P < 0.05). (C) Relative N gene RNA expression in endothelial cells from vesseloids or Calu-3 cells at 3 days post infection with SARS-CoV-2 (MOI = 0.1). (D) Immunofluorescence analysis of cleaved-Caspase-3 (c-Caspase-3) in vesseloids 3 days post infection (MOI = 0.1). The fluorescence intensity per nucleus was quantified. The results are presented as the means ± SEMs.
To corroborate these findings, we analyzed the mRNA levels of the N protein, the nucleocapsid of the virus, in WT-HUVEC vesseloids and in the epithelial cell line Calu-3 at 3 days after SARS-CoV-2 infection. Significantly lower levels were detected in vesseloids than in Calu-3 cells (mean control vesseloid = 1 ± 0 and SARS-CoV-2 vesseloid = 72.41 ± 16.30 vs. mean control Calu-3 = 1 ± 0 and mean SARS-CoV-2 Calu-3 = 9114 ± 9754) (Fig. 1C).
These findings indicate that infection of endothelial cells did not result in efficient viral replication and particles production even in the presence of ACE2.
Subsequently, we examined whether the virus has an impact on endothelial cell viability in vesseloids after 3 days of SARS-CoV-2 infection (MOI = 1) and performed immunostaining for cleaved-Caspase-3 (c-Caspase-3) protein detection. However, cleaved-Caspase-3 expression remained unchanged, regardless of whether the cells were exposed to the virus. These results indicated that the virus did not directly induce endothelial cell death (mean control vesseloid = 0.2162 ± 0 vs. mean SARS-CoV-2 vesseloid = 0.1015 ± 0.02141) (Fig. 1D). This finding further supports the hypothesis that endothelial cells low viral uptake does not allow SARS-CoV-2 virus replication and subsequent induced cell death.
Epithelium-dependent effects of SARS-CoV-2 infection on endothelial cells
To determine the mechanisms of endothelial activation/inflammation, we co-cultured vesseloids with Calu-3 cells. Calu-3 cells were plated in the bottom chamber and infected with SARS-CoV-2 for 4 h (MOI = 1). After several washes, the vesseloids were placed in the upper part of the transwell insert, and cocultures were maintained for 3 days (Fig. 2A). Analysis of RNA expression of the vascular adhesion marker VCAM-1 in the vesseloids revealed significantly greater levels in co-cultured vesseloids than in untreated vesseloids (mean number of SARS-CoV-2 vesseloids = 1.050 ± 0.1212 vs. mean number of SARS-CoV-2 vesseloid cocultures = 11.36 ± 3.536) (Fig. 2B). An increase tendency (p = 0.07) in intercellular adhesion molecule (ICAM-1) mRNA was also observed in co-cultured vesseloids (Sup Fig. 2A). This observation demonstrated cross-talk between infected epithelial Calu-3 cells and endothelial cells even when the cells were included in the 3D vesseloids. This finding may mirror the in vivo situation of endothelial cell activation during SARS-CoV-2 infection.

The inflammatory response of ECs is mediated by epithelial-derived CXCL10. (A) Calu-3 cells were infected with SARS-CoV-2 (MOI = 1) for 4 h and plated at the bottom of the transwell chamber before they were cocultured with vesseloids placed in the upper chamber for 3 days. (B) Relative VCAM-1 mRNA expression in vesseloids infected with SARS-CoV-2 alone or after coculture with Calu-3 cells infected with SARS-CoV-2. Analyze was performed 3 days post infection (MOI = 0.1). The values were normalized according to tubulin mRNA expression. The results are presented as the means ± SEMs (unpaired t test; *P < 0.05). (C) Heatmap of pixel density quantification from the cytokine array assay for selected chemokines and cytokines from vesseloids infected alone or after coculture with Calu-3 cells. SARS-CoV-2 was added to the culture supernatant, which was incubated for 4 h and analyzed 3 days post infection (MOI = 1). (D) Relative CXCL10 mRNA expression in SARS-CoV-2-infected vesseloids alone or after coculture with infected Calu-3 cells or infected Calu-3 cells alone. The data were analyzed 3 days post infection by SARS-CoV-2 (MOI = 0.1). The results are presented as the means ± SEMs (unpaired t test *P < 0.05). (E) CXCL10 concentration in the supernatant of infected vesseloids alone or after coculture with infected Calu-3 cells or infected Calu-3 cells alone. Analyze of the cells was performed 3 days post infection (MOI = 0.1). The results are presented as the means ± SEMs (one-way ANOVA, *P < 0.05).
To determine the molecular mechanism involved, we compared inflammatory cytokine profiles in the supernatants of vesseloids cultured alone or in co-culture with Calu-3 cells infected with SARS-CoV-2 via a cytokine array (R&D Systems). The level of the chemokine CXCL10 strongly increased under co-culture conditions (Fig. 2C). We discovered that CXCL10 mRNA expression was also highly increased in vesseloids co-cultured with infected Calu-3 cells compared to vesseloids cultured alone. Furthermore, the expression of this cytokine was also increased in infected Calu-3 cells cultured alone (mean SARS-CoV-2 vesseloid = 1.050 ± 0.1212 vs. mean SARS-CoV-2 Calu-3 = 212.9 ±290.2 vs. mean SARS-CoV-2 vesseloid coculture = 116.2 ± 89.41) (Fig. 2D). To validate these findings, we analyzed the amount of CXCL10 in the culture media of vesseloids alone, of Calu-3 cells or during coculture. We observed a significant increase in CXCL10 in the culture medium when vesseloids were co-cultured with infected Calu-3 cells (mean number of SARS-CoV-2 vesseloid cocultures = 56,869 ± 47787) (Fig. 2E). For Calu-3 cells infected alone, we detected a slight increase in CXCL10 in the culture medium (mean number of SARS-CoV-2 Calu-3 cells = 1323 ± 1079) (Fig. 2E). When vesseloids were infected with SARS-CoV-2, CXCL10 levels remained unchanged (mean number of SARS-CoV-2 vesseloids = 3.810 ± 0.4424) (Fig. 2E). These data are in agreement with the CXCL10 mRNA expression observed in both vesseloid endothelial cells and infected Calu-3 cells (Fig. 2D). IL6 and IL8 protein concentrations in the culture supernatant were also determined. In Calu-3 cells, IL6 expression was increased but was not further increased when the cells were cocultured with vesseloids. For IL8, no significant further increase was observed above the background level in Calu-3 cells cultured alone or in combination with vesseloids (Sup Fig. 2B). These findings highlight the essential role of CXCL10 in the crosstalk between endothelial cells and the infected epithelium.
Mechanistical insights into the activity of chemokines involved in SARS-CoV-2 infection pathophysiology
To understand the process of endothelial alterations following SARS-CoV-2 infection, we stimulated HUVECs with recombinant CXCL10, the most highly expressed cytokine in our study.
First, endothelial cell permeability was tested using an in vitro permeability assay in which a confluent monolayer of HUVECs was cultured in the upper part of the insert and treated with CXCL10. Carrier or VEGF was used as a negative or positive control respectively. FITC-dextran was then added to the upper part of the transwell plate, after which the FITC-dextran was collected in the bottom chamber, after which the optical density was measured. We observed a significant increase in endothelial permeability in response to CXCL10 and VEGF compared to that in the control group (mean control = 15.17 ± 7.522 vs. mean CXCL10 = 53.13 ± 12.22 vs. VEGFA-A mean = 30.13 ± 4.029; at 60 min) (Fig. 3A).

Epithelial-derived CXCL10 opens inter-endothelial junctions. (A) HUVECs were seeded in the upper well of a transwell chamber and grown to confluency. Cells were treated with 1 µg/ml CXCL10 or 1 µg/ml VEGF-A for 1 h after 25 µg/ml FITC-dextran was added to the upper well. Every 20 min, after the addition of dextran, the fluorescence in the lower compartments was measured. The results are presented as the means ± SEMs (one-way ANOVA, *P < 0.05). (B) For quantification of VE-cadherin junctions, HUVECs were seeded, grown to confluency and treated for 4 h with 1 µg/ml CXCL10 or 1 µg/ml VEGF-A and VE-cadherin, and actin was stained. The results are presented as the mean ± SEM (one-way ANOVA, **P < 0.01). (C) VE-cadherin immunostaining after stimulation for 4 h with 1 µg/ml CXCL10 or 1 µg/ml VEGF-A. Green indicates VE-cadherin, red indicates actin, and blue indicates DAPI.
To decipher the biological mechanisms underlying the increase in permeability, we analyzed endothelial cell‒cell junctions by immunostaining, focusing on the endothelial-specific junction marker VE-cadherin. Similar to VEGF, stimulation with CXCL10 disrupted VE-cadherin continuous localization at endothelial intercellular contacts in confluent HUVEC monolayers, reflecting destabilization of cell‒cell junctions (control mean = 0.1056 ± 0.02289 vs. CXCL10 mean = 0.3584 ± 0.06868 vs. VEGF-A mean = 0.3131 ± 0.06017) (Fig. 3B and C). Destabilization of cellular junctions appears to be a major factor contributing to vascular permeability and is directly controlled by CXCL10.
CXCL10-dependent myeloid recruitment following SARS-CoV-2 infection
Given the impact of CXCL10 on innate immune cell biology and dynamics, but also on immuno-vascular modulation under pathological conditions such as SARS-CoV-2 infection, we investigated its effect on the recruitment of peripheral myeloid cells (Fig. 4). To this end, we first investigated the effect of CXCL10 on the migration of activated human THP1 monocytic cell line derived from a monocytic leukemia patient using a transwell assay approach to determine the ability of THP1 cells to follow a CXCL10 gradient. This directional migration assay revealed a significant monocyte polarization toward the CXCL10 gradient (CXCL10 group mean = 61.33 ± 8.425) in comparison to the control group (control group mean = 16.21 ± 3.951 ) (Supplementary Fig. 3A and 3B). We next investigated whether CXCL10 expressed in the epithelium is also able to induce trans-endothelial cell migration of THP1 cells. To this aim, we overexpressed CXCL10 in Calu-3 epithelial cells using lentiviral infection. The ability of epithelial-derived CXCL10 to allow myeloid cell trans-endothelial migration was confirmed in a three-cell type transwell coculture assay (Fig. 4A–C): Calu-3 cells derived CXCL10 from the bottom chamber significantly increases the migration of THP1 monocytes seeded in the transwell upper chamber through a confluent HUVEC monolayer (CXCL10 group mean = 28.27 ± 6.434) in comparison to control Calu-3 cells (control group mean = 10.87 ± 1.754). To activate cell signaling, CXCL10 binds to its trans-membrane receptor CXCR3. CXCR3 protein was found to be expressed mainly in HUVECs and THP-1 cells, but was expressed at much lower levels in Calu-3 cells evidenced by western blot analysis (Sup Fig. 3). This effect was significantly reduced in the presence of the CXCR3 antagonist SCH 546,738 (CXCL10 + SCH group mean – 19.50 ± 3.162).

Epithelial derived CXCL10 activates endothelial cells and allows myeloid cells trans-endothelial migration through a CXCR3 / Src kinase dependent signaling. (A) Calu-3 cells were seeded at the bottom of the transwell chamber for 48 h before they were cocultured with confluent HUVEC endothelial cells placed in the upper chamber, together with a THP1 monocytes suspension for 24 h. (B) Pictures of activated THP-1 cells which have transmigrate through HUVEC confluent monolayer. The number of transmigrating cells was then counted. (C) Quantification of (B). The results are presented as the means ± SDs (one-way ANOVA; *P < 0.05, ***P < 0.001). (D) Western blot analysis of p-Src, Src, p-ERK and the ERK protein. Vinculin was used as a loading control. (E) Quantification of the data in (D). Bar graphs are presented as the means ± SDs. (F) THP-1 cells were placed in the upper chamber, and migration was stimulated by CXCL10 in the lower chamber. The number of transmigrating cells was then counted after DAPI staining. Treatment with 1 µg/ml CXCL10 alone (blue) or in combination with 6 nM SCH546738 or 1 nM PP2 compared to the control. (G) ELISA dosage of CXCL10 in the supernatant of HUVECs, control scrambled Calu-3 cells, Calu-3 cells transfected with CXCL10 expressing lentivirus alone or in coculture with HUVECs. The results are presented as the means ± SDs (one-way ANOVA; *P < 0.05, **P < 0.01, and ***P < 0.001).
To determine the downstream signaling pathway responsible for this migration, we treated THP1 cells with CXCL10 for various durations and analyzed the downstream substrates by western blot analysis. We focused our analysis on the Src pathway, which was previously reported to be a potent downstream cue of myeloid cell migration16. Src phosphorylation was significantly increased after CXCL10 stimulation in THP-1 cells after 10 min (control mean = 1 ± 0 vs. 10 min mean = 2.5443 ± 1.572) (Fig. 4D and E). As a control, the ERK pathway, which is responsible for cell proliferation and survival in myeloid cells in response to CXCL1017, was also studied and found to be activated, with peak phosphorylation occurring at 15 min. CXCR3 blockade by SCH 546,738 antagonist treatment inhibited THP-1 cell migration (Fig. 4F). The same result was observed when PP2, a Src family kinases inhibitor, was used (control mean = 26.33 ± 2.215 vs. CXCL10 mean = 93.53 ± 9.101 vs. CXCL10 + SCH mean = 43.10 ± 5.734 vs. CXCL10 + PP2 mean = 37.73 ± 8.768) (Fig. 4F). It is to note that CXCL10 epithelio-endothelial mediated cross-talk enhanced CXCL10 production in co-culture: dosage of CXCL10 in the supernatant of HUVECs, Calu-3 control cells, CXCL10 overexpressing Calu-3 cells, alone or co-cultures revealed that native HUVECs produced a low amount of CXCL10 (mean = 1.250 ± 0.207 pg/ml) compared Calu-3-CXCL10 (mean = 197.9 ± 17.49 pg/ml), but that co-culture of these two cell types enhances dramatically CXCL10 production (mean = 331.9 ± 23.08 pg/ml) (Fig. 4G). This will potentialize trans-endothelial migration of immune cells.
Taken together, these results showed that CXCL10 plays a crucial role in the recruitment of myeloid cells via activation of the SRC/MAP kinase pathway. Also, epithelial cell-derived CXCL10 production induces endothelial CXCL10 secretion, which in turn favors immune cell recruitment.
Clinical correlations
To corroborate our results and test whether our in vitro results reflected clinical conditions, we analyzed the serum of two patient subgroups from the COLCOV cohort, each of which presented moderate or severe symptoms following SARS-CoV-2 infection, and quantified a number of circulating cytokines, including not only CXCL10 but also IL6 and IL8, for comparison (Fig. 5A). Circulating CXCL10 levels were significantly increased in patients with SARS-CoV-2 infection (CXCL10 control mean = 9.484 ± 4.197 pg/ml; CXCL10 moderate mean = 208.9 ± 156.5 pg/ml; and CXCL10 severe mean = 199.9 ± 183.6 pg/ml). This was already the case in patients with mild infection. Furthermore, a significant increase in IL6 was also observed, but only in patients with severe infection. Circulating IL8 levels also showed a tendency for increase in sever SARS-CoV-2 infected patients (Fig. 5A). The increased level of circulating cytokines was accompanied by significantly higher levels of sFlt1, VEGF and Angiopoietin, which are markers of endothelial cell activation (Fig. 5B). Unexpectedly, circulating VE-Cadherin and CD31 fragments were significantly decreased in the blood of SARS-CoV-2 infected patients (Fig. 5C). This is different to other chronic inflammatory pathologies which are linked to endothelial activation. One can only speculate about the reason for this difference. Based on our in vitro analysis in which CXCL10 limits VE-Cadherin localization at the cell surface, disrupting endothelial cell-cell junction without affecting its expression level, we hypothesize that the amounts of membrane-bound VE-Cadherin and CD31 may be reduced through internalization or export mechanisms, ultimately reducing the amount available for shedding by the action of metalloproteinases.

Clinically relevant serum samples and bioinformatics data from SARS-CoV-2-infected patients. (A) CXCL10, IL6 and IL8 protein levels in the plasma of SARS-CoV-2-infected patients (severe or moderate) and SARS-CoV-2-noninfected patients (Bordeaux, COLCOV collection). Moderate = acute respiratory syndrome coronavirus 2 without intensive care unit transfer; severe = severe acute respiratory syndrome coronavirus 2 requiring intensive care unit transfer. The results are presented as the mean ± SEM (one-way ANOVA, ***P < 0.001). (B) Representation of Flt1, VEGF and Ang2 levels in the plasma of SARS-CoV-2-infected patients (severe or moderate) and SARS-CoV-2-noninfected patients. (C) Representation of VE-Cadherin and CD31 levels in the plasma of SARS-CoV-2-infected patients (severe or moderate) and SARS-CoV-2-noninfected patients.
To confirm the results from our biological and clinical analyses, we investigated CXCL10 expression in an independent cohort of COVID-19 patients in silico. To this end, we analyzed the differences in CXCL10 expression between SARS-CoV-2 infected patients and control healthy individuals in an RNA-seq cohort (GSE152075) available online, and statistically significant differences were found (Supplementary Fig. 4). CXCL10 expression was highly upregulated in SARS-CoV-2 infected patients compared to controls. We also assessed the association between the expression of CXCL10 and genes well known to be involved in endothelial activation (ICAM-1 and F3) and inflammation (IL8). The expression of these genes was significantly correlated with CXCL10 expression in SARS-CoV-2 infected patients (Supplementary Fig. 4A).
In parallel, we analyzed the expression of genes related to M1 and M2 macrophage signatures in patients with low, medium, or high CXCL10 expression. We observed an increase in the mean Max-Min-normalized expression of the M1 and M2 genes, which was similar to the CXCL10 expression pattern (Supplementary Fig. 4B). We also calculated Pearson correlation coefficients to determine the possible tendency toward a positive correlation between CXCL10 expression and M1/M2 macrophages, representing genes in SARS-CoV-2 patients, compared to those in the control group. These findings are consistent with our in vitro analysis and support the general concept of CXCL10 cross-talk between epithelial and endothelial cells following SARS-CoV-2 infection.
Discussion
In this study, we explored the interaction between SARS-CoV-2 and the vascular endothelium. Our findings revealed that endothelial cell viral uptake is limited and does not result in efficient viral replication and particle production in the presence or absence of ACE2. Instead, we observed indirect endothelial cell activation, which is mediated by CXCL10. Furthermore, CXCL10 also serves as an endothelial cell-derived chemoattractant for myeloid cells. Serum analysis of COVID-19 patients supported this contention since CXCL10 levels were elevated in both moderate and patients with severe COVID-19. This was further supported by the in silico analysis of an independent patient cohort.
Multiple studies have reported conflicting results regarding the role of the vascular endothelium in the pathogenesis of COVID-193,14,18,19.
Analysis of deceased patient tissue seems to reveal the presence of SARS-CoV-2 in endothelial cells14. However, the ACE2 receptor, which has been demonstrated to be determinant for viral entry, was detected only in pericytes of the brain, heart and pancreatic vessels and not in endothelial cells, or at extremely low levels, which lower the possibility of significant virus entry in ECs20. Nevertheless, transcriptomic analysis revealed the presence of ACE2 in 20 different tissue types and in several types of endothelial cells, including the Fallopian tube, thyroid, parathyroid gland, adrenal gland, pancreas, and heart21. This supposition has also been supported by claims of ACE2 expression in human pulmonary artery endothelial cells. However, a deep transcriptomic analysis via scRNAseq and epigenomic data indicated that human ECs from tissues or cell lines do not express ACE210. This latter analysis is consistent with our findings because we were also unable to detect the presence of ACE2 in all the human EC lines we tested, including human coronary endothelial cells. Thus, in our experience, ECs do not express ACE2.
Nevertheless, SARS-CoV-2 may enter ECs via an unidentified receptor. ACE2 independent SARS-Cov-2 cellular entry mechanisms have been described and the existence of an alternative SARS-CoV-2 receptor has been recently reported22. For this reason, we examined whether SARS-CoV-2 could infect human endothelial cells and replicate efficiently and whether the cells expressed ACE2. To more closely mimic the in vivo architecture of blood vessels, we utilized a vesseloid model, which we previously described15, in our experiments.
In cells that did not express ACE2, SARS-CoV-2 was unable to enter and replicate efficiently in ECs even when it was present in a 3D vascular architecture. We then investigated whether the expression of ACE2 in ECs allows virus entry and replication. However, even under these conditions, no efficient virus replication was observed, although virus entry increased to some extent. Taken together, these findings did not support the idea of direct viral interaction with endothelial cells.
Thus, ECs are likely activated indirectly via a paracrine mechanism and/or inflammatory molecules present in the blood. It is also unlikely that the recently described alternative receptor TMEM 106B is overexpressed in ECs since this receptor was not expressed in the ECs we studied and because no efficient virus replication was found in these cells22.
We demonstrated cross-talk between epithelial and endothelial cells, with CXCL10 playing a major role. CXCL10 was overexpressed in both epithelial cells and endothelial cells in the presence of SARS-CoV-2, which was confirmed by the protein concentration in the culture medium. In the absence of viral infection, only low levels of CXCL10 were detected in the medium. CXCL10 is known to interact with CXCR3A and B23,24, and a plethora of biological functions, such as immune regulation, tumor cell invasion25 and angiogenesis26, have been described for this chemokine. Therefore, in parallel, we examined CXCR3 expression in ECs and epithelial cells and found that CXCR3 was indeed expressed in both cell types.
We next examined the function of CXCL10 in the activated endothelium. CXCL10-mediated disruption of EC cell junctions is dependent on VE-cadherin, which is associated with increased EC permeability27. Many studies have demonstrated the destabilization of VE-cad junctions during infection with SARS-CoV-2; one study even suggested that this protein could be an effective biomarker for detecting long-term SARS-CoV-2 infections28. This finding is supported by the observation that culture of plasma from COVID-19 patients disrupted the EC junctions of primary human pulmonary microvascular endothelial cells by affecting VE-cadherin expression29.
Proteome analysis of the plasma of a cohort of early hospitalized patients infected with SARS-CoV-2 revealed 27 proteins, which included complement factors, the coagulation system, inflammation modulators, and proinflammatory factors upstream and downstream of IL6. CXCL10 was not detected in this study30. Endothelial cell damage caused by SARS-CoV-2 is the result of inflammation via an indirect mechanism. This finding is supported by studies demonstrating the adhesion of monocytes to endothelial cells during SARS-CoV-2 infection31. This finding is in line with the increase in monocyte migration and p-Src activation after CXCL10 stimulation shown in our study, which favors EC-monocyte/macrophage cross-talk and is consistent with the findings of other studies that highlight a role for CXCL10 during SARS-CoV-2 infection32. In another publication, a direct effect of SARS-CoV-2 on monocytes was proposed33. Monocytes were exposed to inactivated SARS-CoV-2, and the cytokine/chemokine profile revealed significantly greater levels of IL6, TNF-α, CXCL10, CXCL9 and CXCL11. This interaction may represent an alternative target for SARS-CoV-2 infection that contributes to cytokine/chemokine modulation in addition to the cross-talk we demonstrated in our work.
Our mechanistic hypothesis implicating CXCL10 as a major chemokine regulating epithelial-endothelial interactions during SARS-CoV-2 infection was reinforced by inhibitor studies using SCH54738 and PP2. The former inhibits the binding of CXCL10 to CXCR3 and SRC phosphorylation.
A CXCL10-independent mechanism has also been proposed. The complement system has been shown to contribute significantly to vesseloid inflammation after SARS-CoV-2 infection. Qin Z et al.34 analyzed the effect of infection in various models, including a murine model and a primate model, and analyzed postmortem biopsies using RNAscope or electron microscopy. Their data revealed an increase in complement deposition in the lungs in murine models and inflammation. Proteomic analysis of the blood of COVID-19 patients revealed the presence of complement factor D (CFD) and abnormalities in D-dimer and fibrinogen/fibrin degradation products (FDP). These findings suggest that the complement system plays a central role in SARS-CoV-2 infections. Complement system activation could have deleterious effects after 2 to 3 weeks following infection, potentially leading to acute respiratory distress syndrome (ARDS)35. Finally, we investigated chemokine and cytokine expression in the blood circulation of COVID-19 patients. This analysis confirmed highly significant CXCL10 overexpression in patients with mild and severe COVID-19. This expression pattern was different from that of IL6 or IL8. Not minoring the impact of alternative cytokines such as IL6 or IL8, this highlights the potential central role of CXCL10 in COVID-19 pathogenesis. Thus, CXCL10 inhibition could constitute a potential suitable therapeutic strategy for the treatment of COVID-19. These results were reinforced by the analysis of an independent patient cohort. Endothelial cell inflammatory markers were coexpressed with CXCL10 as well as M1 or M2 macrophages on the basis of a biased selection of monocyte/macrophage markers. Both M1 and M2 markers were coexpressed, which points to the recruitment of both M1 and M2 macrophages. This, however, does not take into account the time-dependent evolution of the disease. Indeed, the proportion of M1- or M2- recruited macrophages should vary with time, with initial M1 macrophage activation followed by M2 macrophage recruitment.
Taken together, our results are of quadruple significance. First, our findings suggest that a direct effect of SARS-CoV-2 is highly unlikely and that it favors indirect cross-talk between ECs, epithelial cells and monocytes. In our study, the infected epithelium was the prime target of infection, which triggered sequential activation of endothelial cells and monocytes. Second, in our experience, the major player in this sequential interaction is CXCL10, which expression in epithelial cells catalysis its expression in endothelial cells and promotes the attraction of monocytes. Third, CXCL10 is able to disrupt EC junctions, thus allowing transmigration of inflammatory cells across the endothelial cell barrier. Fourth, these effects can be inhibited by CXCR3 blockade and thus may constitute an interesting therapeutic strategy.
Novelty and limitations of the study
CXCL10 has been described in COVID-1932. However, there are conflicting results about the cellular interactions and mechanisms. Most of the publications are dealing with CXCL10 in a general inflammatory context which is different from our study. One paper has even isolated endothelial cells from aged infected mice and show endothelial cell activation but with no significant increase of CXCL10 (despite the claim in the abstract)36.
The novelty of our paper relies (1) in the demonstration that a double CXCL10 loop is involved between epithelial cells, endothelial cells and immune cells, and (2) that, for this demonstration, a 3D vascular model (“vesseloid model”) developed in our laboratory14 was used, besides classical 2D models. This validates our contention in a model that mimics a vascular structure in a 3D configuration. Finally, these data are supported by analysis of samples from patients with mild or sever SARS-CoV-2 infection and in silico analysis. Our data may also have a broader significance in the context of other infections where the vasculature is involved or in inflammatory vascular disease.
A limitation of the study is the absence of in vivo results using rodents, guinea pigs or larger animal such as monkeys. However, we have used instead complex in vitro models such the “vesseloid” model which mimics the configuration of a blood vessel. Another limitation is that we performed some of the mechanistic experiments with recombinant proteins and not with endogenously produced protein. However, the expression data clearly demonstrate a double CXCL10 cytokine loop, and the experiments related to molecular mechanism of CXCL10, using recombinant protein, are, albeit confirmatory, nevertheless essential to substantiate the whole contention.
Materials and methods
Viruses and cell lines
Vero-E6 (ATCC, CRL-1586) and Calu-3 cells (ATCC, HTB-55) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal calf serum and gentamicin (50 µg/mL) at 37 °C in a humidified 5% CO2 incubator. HUVECs (Promocell, C-12203), HUAECs (Promocell, C-12202), and HCAECs (Promocell, C-12221) were maintained in endothelial cell growth medium 2 (EGM2; C-22111, PromoCell) in a cell culture dish coated with 0.5% gelatin solution. Human monocytic THP-1 cells (ATCC, TIB-202) were cultured in RPMI-1640 medium (Life Technologies) supplemented with 10% FBS, 2 mM L-glutamine and 1% penicillin and streptomycin. THP-1 cells macrophage differentiation was performed using 10 mM phorbol 12-myristate 13-acetate (PMA) treatment for 24 h.
The SARS-CoV-2 strain BetaCoV/France/IDF0372/2020 was supplied by the National Reference Centre for Respiratory Viruses hosted by the Pasteur Institute through the European Virus Archive (EVAg platform). The work with infectious SARS-CoV-2 was performed in a Class III Biosafety Cabinet under BSL-3 conditions at the UB’L3 facility (TBMcore, University of Bordeaux).
Vesseloid production
Vesseloids are engineered blood vessels. They were produced using a microfluidic technology adapted from37,38. In our seminal work15, we used a coculture of HUVECs and smooth muscle cells to achieve vesseloid contractility. Here, HUVECs were used only for vesseloid production, which was carried out according to the same procedure. Briefly, by using a 3D printer coextrusin device, we produced hollow alginate fibers whose inner walls were coated with ECM (Matrigel) and filled with HUVECs. Cells spontaneously self-organize to create a lumen within 24 h.
Virus production
The SARS-CoV-2 strain was produced by infecting Vero E6 cells with a multiplicity of infection of 10− 2 PFU and then incubating the cells at 37 °C in a humidified CO2 incubator until a cytopathic effect was observed (approximately 72 h). The culture supernatant was clarified by centrifugation (5 min at 1500 rpm), and aliquots were stored at – 80 °C. All the viral stocks were sequenced to confirm that no mutations were detected during culture, and Sanger sequencing was used to determine the full-length spike sequence; moreover, whole-genome sequencing was performed with Oxford Nanopore technology. Stock titers were determined by adding serial dilutions to 2 × 104 Vero E6 cells in supplemented DMEM in a 96-well plate. Eight replicates were performed. The plates were incubated at 37 °C, after which the cytopathic effects were examined. Cytopathic effects were quantified using the CellTox Green cytotoxicity assay (Promega) according to the manufacturer’s instructions and a Victor Nivo reader (Perkin-Elmer). The TCID50 per ml was calculated according to the method of Reed and Muench39. The viral titer expressed in PFU per ml was mathematically converted from the TCID50 per ml determination using the Poisson equation as follows: PFU/ml = – ln 0.5 * TCID50/ml.
Infection of vesseloid and Calu-3 epithelial cells
Calu-3 cells were seeded in a 24-well plate at 100,000 cells/well the day before infection. Infection was performed with SARS-CoV-2 at a multiplicity of infection of 0,1. Vesseloids were placed on cell culture inserts and inserted into wells containing SARS-CoV-2-infected Calu-3 cells until infection was stopped.
Vesseloid and supernatant processing
RNA extraction from vesseloids was performed using TRI Reagent (TR118, Euromedex). Vesseloids were first washed with 500 µL of DMEM and then incubated with 1 mL of ReLeSR (100–0484, StemCell) for 5 min to dissolve the alginate. The cells were pelleted by centrifugation (5 min at 1,500 rpm) and treated with 600 µL of TRI Reagent for 10 min. For microscopic observation, the vessels were fixed with 4% paraformaldehyde (PFA) for 20 min and washed with DMEM.
Five hundred microliters of culture supernatant were either inactivated with 0.5% Triton for cytokine measurement or with lysis buffer for RNA extraction according to the manufacturer’s recommendations (High Pure Viral RNA Kit, Roche).
Quantification of SARS-CoV-2 RNA by RT‒qPCR
For RT‒qPCR quantification of viral RNA, total RNA was isolated using the High Pure Viral RNA Kit (Roche) according to the manufacturer’s instructions. Viral RNA was quantified using a GoTaq 1-Step RT‒qPCR Kit (Promega). SARS-CoV-2 N gene RNA was amplified using forward primers (N-gene F: CGCAACAGTTCAAGAAATTC; N-gene R: CCAGACATTTTGCTCTCAAGC). Serial dilutions of SARS-CoV-2 RNA were used to construct a standard curve. The amplification program began with an RT step for 15 min at 50 °C, followed by a denaturation step for 10 min at 95 °C, 10 s at 95 °C, 10 s at 60 °C, and 10 s at 72 °C (40 cycles). The melting curve was obtained by increasing the temperature from 60 °C to 95 °C at 0.5 °C/s. Data are mentioned as “Ngene expression (equivalent PFU)” in regards to the standard curve comparison with known PFU values.
Quantification of gene expression by rt‒qpcr
Total RNA was isolated from cells using TRI Reagent (TR118, Euromedex) according to the manufacturer’s instructions. 1 µg of total RNA was reverse transcribed into complimentary DNA (cDNA) using a high-capacity cDNA reverse transcription kit (4368814, Applied Biosystems). cDNAs were amplified using specific primers for the genes of interest. Tubulin (TUB R: TAGAGCTCCAGCAGGATT; TUB F: GAGTGCATCTCCATCCACGC) was used as an internal control. We applied real-time PCR using Eurobiogreen qPCR Mix (GAEMMX02H 0T, Eurobio) for Cxcl10 (CXCL10 R: GGCCTTCGATTCTGGATTCA; CXCL10 F: GCCAATTTTGTCCACGTCGTTG); Vcam-1 (VCAM-1 R: ACATTCATATACTCCCGCATCCTTC; VCAM-1 F: CGTCTTGGTCAGCCCTTCCT); Icam-1 (ICAM-1 R: CCCATTATGACTGCGGCTGCTA; ICAM-1 F: AGGCCACCCCAGAGGACAAC); Il6 (IL6 R: GCAAGTCTCCTCATTGAATCC; IL6 F: GGCACTGGCAGAAAACAACC); Il8 (IL8 R: AACCCTCTGCACCCAGTTTTC; IL8 F: TGCAGCTCTGTGTGAAGGTG); Ngene (N-gene R CCAGACATTTTGCTCTCAAGC; N-gene F CGCAACAGTTCAAGAAATTC). Real-time PCR was performed on an AriaMx Real-time PCR System using Agilent Aria 1.71 software (Agilent).
Cytokine array assay
Cytokine profiling was performed using a Proteome Profiler Array Human (XL) Cytokine Array Kit (ARY022B, R&D Systems), which detects 105 human cytokines simultaneously. We used cell culture supernatants collected from infected vesseloids and cocultures (SARS-CoV-2-infected Calu-3 cells and vesseloids) according to the manufacturer’s instructions. The arbitrary values of cytokine pixels were calculated for each dot plot, normalized to the reference spots and measured using ImageJ software (2.14.0/1.54f).
Luminex dosages
Blood biomarkers were measured using Luminex technology and ELISA according to the manufacturers’ instructions. Angiopoietin-2, VEGFR1, and CD31 were measured using a custom 3Plex (Biotechne); CXCL10, IL8, IL6, and VEGF were measured using a custom 4plex (Biotechne); and CD142 (ab108903, Abcam) and VE-Cadherin (DCADV0, Biotechne) were measured using commercial ELISA. All the measurements were performed on the CHU hospital research platform (PARS) on samples stored at – 80 °C in a centralized biobank (Bordeaux Biothèques Santé Centre de Ressources Biologiques, Bordeaux University Hospital) blinded to any other data.
Western blot
The cells were washed twice with PBS and dissolved in lysis buffer (10 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.5% NP-40; 1% Triton X-100; and 1 mM EDTA supplemented with protease and phosphatase inhibitor cocktails (Roche)). The protein concentration was quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific). The cell lysates were resuspended in Laemmli buffer 4 × (62.5 mM Tris (pH 6.8), 10% glycerol, 2.5% SDS, and 2.5% β-mercaptoethanol). All the samples were boiled for 5 min, subjected to electrophoresis and transferred. The proteins were electroblotted on nitrocellulose membranes. The membranes were incubated with intercept buffer (927-70001, LICOR) for 1 h and probed overnight at 4 °C with primary antibodies. The following antibodies were used: anti-ACE2 (38241 S, Cell Signaling) 1:1000; anti-SRC (Tyr416) (6943 S, Cell Signaling) 1:1000; anti-SRC (2109 S, Cell Signaling) 1:1000; anti-p-p44/42 MAPK (Erk1/2) (4370 L, Cell Signaling); anti-p44/42 MAPK (Erk1/2) (9102 S, Cell Signaling) 1/1000; anti-p-AKT (Ser473) (9271 S, Cell Signaling); anti-AKT (9272 S, Cell Signaling); and anti-vinculin (VLN01) (11394493, Invitrogen). Following washes, secondary antibodies goat anti-rabbit IR Dye 680CW (926-68021, LI-COR Biosciences), goat anti-rabbit IR Dye 800CW (926-32211, LI-COR Biosciences) and goat anti-mouse IR Dye 800CW (926-32210, LI-COR Biosciences) were incubated at 1:1000 for 2 h at RT, membranes were washed and imaged using Odyssey infrared imaging system (LI-COR Biosciences, Nebraska, US). Analysis was performed using Image Studio Lite 5.2 software.
Immunofluorescence
Cells were seeded on glass coverslips and fixed for 15 min with 4% paraformaldehyde at room temperature. The cells were permeabilized for 5 min with 0.5% Triton X-100, washed with PBS and incubated for 1 h with blocking buffer (PBS containing 0.5% Triton X-100 and 5% BSA) at room temperature. The cells were incubated with primary antibodies against VE-Cadherin (D87F2) (2500 S; Cell Signaling) diluted 1:500 in blocking buffer for 1 h at room temperature, washed with PBS, and incubated with secondary antibodies conjugated to Alexa Fluor 488-AffiniPure Donkey Anti-Rabbit IgG (H + L) (711-545-152; Jackson ImmunoResearch) and phalloidin (diluted in blocking buffer). DAPI (1:1000) was used to label the nuclei (10374168, Fisher Scientific). Coverslips were mounted using Prolong Gold antifade reagent (11559306, Fisher Scientific).
Vesseloids were fixed in 4% PFA overnight at 4 °C. Then, the vesseloids were cut into 1 cm long sections. The cells were permeabilized at room temperature in Dulbecco’s modified Eagle’s medium (DMEM; P04-05545; PAN Technology) supplemented with 0.1% Triton X-100 and incubated with 1% BSA and 2% SVF in DMEM for 1 h at 4 °C under agitation. Vesseloids were incubated with a primary antibody against cleaved-Caspase-3 (Asp175) (9661; Cell Signaling) 1:200 at 4 °C overnight with agitation. The vesseloids were then washed with DMEM 3 times for 15 min and incubated with the secondary antibodies Alexa Fluor 488-AffiniPure Donkey Anti-Rabbit IgG (H + L) (711-545-152, Jackson ImmunoResearch) 1/200 and DAPI 1/500 diluted in blocking buffer for 2 h at 4 °C with agitation.
Vesseloid fragments were fixed in a cellview cell culture dish (627860, Greiner bio-One) with 1% low agarose (BP160500, Fisher Scientific) and covered with DMEM.
Imaging
Confocal images of vesseloids were acquired either with an inverted Nikon Eclipse Ti microscope and acquisition software (NIS-Element, NIS) or with a Leica DFC7000 and acquisition software (Leica Las X). Image analysis was performed using ImageJ software (2.14.0/1.54f).
Permeability assay
HUVECs were cultured on 6-well transwell insert plates until 100% confluence. The cells were treated for 1 h with 1 µg/ml recombinant human CXCL10 protein (300 − 12, Gibco). FITC-conjugated dextran was administered to the upper compartments of the inserts at 25 µg/ml. Every 20 min after dextran addition, the fluorescence signal in the lower compartments was measured using a fluorescence microplate reader (Infinite 200 PRO, Tecan) at an excitation wavelength of 485 nm and an emission wavelength of 538 nm.
Migration assay
For chemotactic migration assays, 8.0 μm polycarbonate membrane Transwell inserts were used in 6-well plates (Corning). 25.000 activated primary THP1 cells were plated in 500 µL of serum-free RPMI medium in the top chambers. Then, the bottom chambers were filled with 2 ml of serum-free RPMI supplemented with 1 µg/ml recombinant human CXCL10 protein (300 − 12 Gibco) or VEGF-A (R&D Systems). The cells were cultured overnight at 37 °C and 5% CO2, fixed with 4% PFA for 15 min and stained with DAPI. Top-chamber cells were striped using a cotton swab, wells were then washed with PBS, and 10 images per well were acquired at 10x magnification. The number of migrating cells per field was semi-automatically counted using ImageJ software (2.14.0/1.54f). For inhibitor assays, cells were seeded in the top chamber, and 6 nM SCH 546,738 (HY-10017, MedChemExpress) or 10 nM PP2 (#73901, Cell Signaling) was added.
Trans-endothelial migration assay
Calu-3 CXCL10 overexpressing cells were generated using lenti-viral particles (RC203141L1V) according to the manufacturer protocol (Origene). Following antibiotics selection, polyclonal population of Calu-3 were expended.
For the 3 cell types coculture migration assay, 50.000 Calu-3-CXCL10 or control Calu-3-Scamble cells were seeded in the bottom chamber of a 24 well plate. On the same day, in a second 24 well plates, 50.000 HUVECs were seeded in the upper part of 3.0 μm polycarbonate membrane Transwell insert and cultured for 48 h in order to form an endothelial confluent monolayer. After 48 h, HUVECs covered inserts are transferred in the Calu-3 cultured 24 well plates and 25.000 THP1-GFP cells were seeded in the upper chamber of the transwell and transmigrating monocytes were analyzed after 24 h. In order to only analyze trans-migrating cells, top-chamber cells were striped using a cotton swab, wells were then washed with PBS, and 10 images per well were acquired at 10x magnification. The number of migrating cells per field was semi-automatically counted using ImageJ software (2.14.0/1.54f). For inhibitor assays, cells were seeded in the top chamber together with the CXCR3 antagonist SCH 546,738 at 6 nM (HY-10017, MedChemExpress).
CXCL10 ELISA assay
500.000 HUVECs, Calu-3-Scramble or Calu-3-CXCL10 for monocellular type conditioned medium production were plated in 10 cm diameter culture dishes in ECGM2 complete medium (Promocell). For coculture conditioned media, 500.000 HUVECs were seeded with 500.000 Calu-3-Scramble or Calu-3-CXCL10 in 10 cm diameter culture dishes in ECGM2 complete medium (Promocell). For all conditions, cells were cultured overnight in complete medium, then washed with PBS and starved in ECBM2 (Promocell), and cultured for 48 h. Conditioned media were then harvested, centrifuged, and supernatant free of cellular debris were collected. 100 µl of supernatant were subjected to CXCL10 ELISA assay (DY266; R&D Systems) according to the manufacturer protocol. Optical Density were measured at 450 nm using SpectraMax plate reader (Molecular Devices).
Bioinformatic analysis
To study the expression of the Cxcl10 gene in SARS-CoV-2 patients, we analyzed publicly available RNA-seq datasets retrieved from the Gene Expression Omnibus (GEO) repository (GSE152075). The dataset contains gene expression profiles from 484 individuals (430 patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and 54 controls). Differential expression analysis was performed to compare the expression between the COVID-19 and control groups using the R package DESeq2 v1.42.040. For further analysis, patients were divided into three groups according to the expression of Cxcl10: low, medium, and high. Correlations between gene expression patterns were estimated with the Pearson correlation method. The coefficients representing the M1 and M2 macrophage signatures were obtained by calculating the mean of the min–max normalized expression values of the genes used for the analysis among all individuals in a group.
Statistical analysis
For continuous variables, the data are presented as the mean ± SD. Differences were considered significant if P was less than 0.05. Between-group comparisons were performed with the Mann‒Whitney U test or t test, depending on the sample size for continuous variables. In cases where more than 2 groups were compared, 1-way ANOVA was performed, followed by Tukey’s multiple comparison test. All analyses were performed using Prism 6.0 software (GraphPad).
Data availability
No large-scale data (transcriptomic, proteomic, etc.) produced by us were included in this article and thus not deposited in an online repertoire. In silico analysis, included in our article, was performed on publicly available datasets. The datasets used and/or analyzed during the current study are available in a supplementary data file and from the corresponding author on reasonable request.
References
Lai, C. C., Shih, T. P., Ko, W. C., Tang, H. J. & Hsueh, P. R. Severe acute respiratory syndrome coronavirus 2 (SARSCoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int. J. Antimicrob. Agents. 55 (3), 105924 (2020).
Copin, M. C. et al. Time to consider histologic pattern of lung injury to treat critically ill patients with COVID-19 infection. Intensive Care Med. 46 (6), 1124–1126. https://doi.org/10.1007/s00134-020-06057-8 (2020). Epub 2020 Apr 23. PMID: 32328726; PMCID: PMC7178098.
Su, H. et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 98 (1), 219–227 (2020). Epub 2020 Apr 9. PMID: 32327202; PMCID: PMC7194105.
Cipolloni, L. et al. Preliminary post-mortem COVID-19 evidence of endothelial injury and factor VIII hyperexpression. Diagnostics (Basel). 10 (8), 575. https://doi.org/10.3390/diagnostics10080575 (2020). PMID: 32784826; PMCID: PMC7460315.
Birnhuber, A. et al. Between inflammation and thrombosis: endothelial cells in COVID-19. Eur. Respir J. 58 (3), 2100377 (2021). PMID: 33958433; PMCID: PMC8112008.
Karki, R. et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184 (1), 149–168e17 (2021). Epub 2020 Nov 19.
Kása, A., Csortos, C. & Verin, A. D. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers. 3 (1–2), e974448. https://doi.org/10.4161/21688370.2014.974448 (2015). PMID: 25838980; PMCID: PMC4372017.
Scialo, F. et al. ACE2: the major cell entry receptor for SARS-CoV-2. Lung 198 (6), 867–877. https://doi.org/10.1007/s00408-020-00408-4 (2020). Epub 2020 Nov 10. PMID: 33170317; PMCID: PMC7653219.
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in Understanding SARS pathogenesis. J. Pathol. 203 (2), 631–637. https://doi.org/10.1002/path.1570 (2004). PMID: 15141377; PMCID: PMC7167720.
McCracken, I. R. et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation 143 (8), 865–868 (2021). Epub 2021 Jan 6. PMID: 33405941; PMCID: PMC7899720.
Duan, T. et al. ACE2-dependent and -independent SARS-CoV-2 entries dictate viral replication and inflammatory response during infection. Nat. Cell. Biol. 26 (4), 628–644. https://doi.org/10.1038/s41556-024-01388-w (2024). Epub 2024 Mar 21.PMID: 38514841.
Eberhardt, N. et al. SARS-CoV-2 [preprint]nfection [preprint]riggers [preprint]ro-atherogenic [preprint]nflammatory [preprint]esponses [preprint]n human coronary vessels. BioRxiv. https://doi.org/10.1101/2023.08.14.553245. (2023).
Delorey, T. M. et al. Leney-Gre Nat. ;595(7865):107–113. doi: https://doi.org/10.1038/s41586-021-03570-8. (2021). Epub 2021 Apr 29. PMID: 33915569; PMCID: PMC8919505.
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395 (10234), 1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5 (2020). Epub 2020 Apr 21. PMID: 32325026; PMCID: PMC7172722.
Andrique, L. et al. A model of guided cell self-organization for rapid and spontaneous formation of functional vessels. Sci. Adv. 5 (6), eaau6562. https://doi.org/10.1126/sciadv.aau6562 (2019). PMID: 31206014; PMCID: PMC6561743.
Recordon-Pinson, P. et al. A simple and fast method to sequence the full-length Spike gene for SARS-CoV-2 variant identification from patient samples. COVID 1, 337–344 (2021).
Arrieta, V. A. et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat Cancer. 2(12):1372–1386. https://doi.org/10.1038/s43018-021-00260-2. (2021). Epub 2021 Nov 29. Erratum in: Nat Cancer. 2022;3(3):373. PMID: 35121903; PMCID: PMC8818262.
Khaddaj-Mallat, R. et al. SARS-CoV-2 deregulates the vascular and immune functions of brain pericytes via Spike protein. Neurobiol. Dis. 161, 105561. https://doi.org/10.1016/j.nbd.2021.105561 (2021). Epub 2021 Nov 13. PMID: 34780863; PMCID: PMC8590447.
Rauti, R. et al. Effect of SARS-CoV-2 proteins on vascular permeability. Elife 10, e69314. https://doi.org/10.7554/eLife.69314 (2021). PMID: 34694226; PMCID: PMC8545399.
Muhl, L. et al. The SARS-CoV-2 receptor ACE2 is expressed in mouse pericytes but not endothelial cells: implications for COVID-19 vascular research. Stem Cell. Rep. 17 (5), 1089–1104 (2022). Epub 2022 Apr 21. PMID: 35452595; PMCID: PMC9022216.
Hikmet, F. et al. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 16 (7), e9610. https://doi.org/10.15252/msb.20209610 (2020). PMID: 32715618; PMCID: PMC7383091.
Baggen, J. et al. TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry. Cell 186 (16), 3427–3442e22. https://doi.org/10.1016/j.cell.2023.06.005 (2023). Epub 2023 Jul 7. PMID: 37421949; PMCID: PMC10409496.
Boyé, K. et al. The role of CXCR3/LRP1 cross-talk in the invasion of primary brain tumors. Nat. Commun. 8 (1), 1571. https://doi.org/10.1038/s41467-017-01686-y (2017). PMID: 29146996; PMCID: PMC5691136.
Koper, O. M., Kamińska, J., Sawicki, K. & Kemona, H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Adv Clin Exp Med. 27(6):849–856. https://doi.org/10.17219/acem/68846. (2018). PMID: 29893515.
Karin, N. & Razon, H. Chemokines beyond chemo-attraction: CXCL10 and its significant role in cancer and autoimmunity. Cytokine 109, 24–28. https://doi.org/10.1016/j.cyto.2018.02.012 (2018). Epub 2018 Feb 12. PMID: 29449068.
Gao, N. et al. CXCL10 suppression of hem- and lymph-angiogenesis in inflamed Corneas through MMP13. Angiogenesis 20 (4), 505–518. https://doi.org/10.1007/s10456-017-9561-x (2017). Epub 2017 Jun 16. PMID: 28623423; PMCID: PMC5702464.
Giannotta, M., Trani, M. & Dejana, E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 26(5):441 – 54. https://doi.org/10.1016/j.devcel.2013.08.020. (2013). PMID: 24044891.
Bouillet, L. et al. Molecular mechanisms of endothelialitis in SARS-CoV-2 infection: evidence for VE-Cadherin cleavage by ACE2. Int. J. Mol. Sci. 24 (15), 12525. https://doi.org/10.3390/ijms241512525 (2023). PMID: 37569899; PMCID: PMC10419376.
Michalick, L. et al. Plasma mediators in patients with severe COVID-19 cause lung endothelial barrier failure. Eur. Respir J. 57 (3), 2002384. https://doi.org/10.1183/13993003.02384-2020 (2021). PMID: 33154030; PMCID: PMC7651836.
Messner, C. B. et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell. Syst. 11 (1), 11–24e4. https://doi.org/10.1016/j.cels.2020.05.012 (2020). Epub 2020 Jun 2. PMID: 32619549; PMCID: PMC7264033.
Ma, Z. et al. A human pluripotent stem cell-based model of SARS-CoV-2 infection reveals an ACE2-independent inflammatory activation of vascular endothelial cells through TLR4. Stem Cell. Rep. 17 (3), 538–555. https://doi.org/10.1016/j.stemcr.2022.01.015 (2022). Epub 2022 Feb.
Callahan, V. et al. The pro-inflammatory chemokines CXCL9, CXCL10 and CXCL11 are upregulated following SARS-CoV-2 infection in an AKT-Dependent manner. Viruses 13 (6), 1062. https://doi.org/10.3390/v13061062 (2021). PMID: 34205098; PMCID: PMC8226769.
Cvetkovic, J. et al. Human monocytes exposed to SARS-CoV-2 display features of innate immune memory producing high levels of CXCL10 upon restimulation. J. Innate Immun.. https://doi.org/10.1159/000535120 (2023). Epub ahead of print. PMID: 37989107.
Qin, Z. et al. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics 11 (16), 8076–8091. https://doi.org/10.7150/thno.61810 (2021). PMID: 34335981; PMCID: PMC8315069.
Kawakami, E. et al. Complement factor D targeting protects endotheliopathy in organoid and monkey models of COVID-19. Cell. Stem Cell. 30 (10), 1315–1330e10. https://doi.org/10.1016/j.stem.2023.09.001 (2023). PMID: 37802037; PMCID: PMC10575686.
Tsumita, T. et al. Viral uptake and pathophysiology of the lung endothelial cells in age-associated severe SARS-CoV-2 infection models. Aging Cell. 23 (2), e14050. https://doi.org/10.1111/acel.14050 (2024). Epub 2023 Dec 14. PMID: 38098255; PMCID: PMC10861199.
Alessandri, K. et al. A 3D printed microfluidic device for production of functionalized hydrogel microcapsules for culture and differentiation of human Neuronal Stem Cells (hNSC). Lab Chip. 16(9):1593 – 604. https://doi.org/10.1039/c6lc00133e (2016). PMID: 27025278.
Le Maout, V. et al. Role of mechanical cues and hypoxia on the growth of tumor cells in strong and weak confinement: a dual in vitro-in Silico approach. Sci. Adv. 6 (13), eaaz7130. https://doi.org/10.1126/sciadv.aaz7130 (2020). PMID: 32232163; PMCID: PMC7096162.
Reed, L. J. & Muench, H. A simple method of estimating 50% endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Love, M. I., Huber, W. & Anders, S. Moderated Estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. https://doi.org/10.1186/s13059-014-0550-8 (2014). PMID: 25516281; PMCID: PMC4302049.
Acknowledgements
We thank Estelle Coupeau BTS students for her participation during her internship. We thank the Bordeaux Imaging Center (BIC) for imaging support and the TBMCore facility for flow cytometry support. The 3D vesseloids were obtained in collaboration with the VoxCell Organoid facility, and SARS-CoV-2 infection was performed at the UB’L3 facility. These two laboratories are affiliated with the CNRS-INSERM and Bordeaux University – UAR TBMCore 2427-US005.
Author information
Authors and Affiliations
Contributions
L.C. and A.B. participated in conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation and writing/original draft preparation; M.L.B., A.B.M. and I.P. were involved in data curation, formal analysis and methodology; P.R.P. and M.S. were involved in data curation and formal analysis; N.P. was involved in tool development; M.L.A. contributed with ideas and general input; F.B. gave advice clinical; GR was involved in methodology and funding acquisition; L.A. was involved in methodology, funding acquisition, review & editing; T.L. participated in investigation; P.N. gave methodological advice and provided intellectual input; T.M. participated in formal analysis, methodology, investigation and supervised the project; A.B. acquired funding and supervised the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Laura, C., Marie-Lise, B., Patricia, RP. et al. CXCL10-dependent epithelial-vascular cross-talk for endothelial activation following SARS-CoV-2 infection. Sci Rep 15, 21129 (2025). https://doi.org/10.1038/s41598-025-08329-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-08329-z
