
Abstract
Understanding disease progression is crucial for detecting critical transitions and finding trigger molecules, facilitating early diagnosis interventions. However, the high dimensionality of data and the lack of aligned samples across disease stages have posed challenges in addressing these tasks. We present a computational framework, Gaussian Graphical Optimal Transport (GGOT), for analyzing disease progressions. The proposed GGOT uses Gaussian graphical models, incorporating protein interaction networks, to characterize the data distributions at different disease stages. Then we use population-level optimal transport to calculate the Wasserstein distances and transport between stages, enabling us to detect critical transitions. By analyzing the per-molecule transport distance, we quantify the importance of each molecule and identify trigger molecules. Moreover, GGOT predicts the occurrence of critical transitions in unseen samples and visualizes the disease progression process. We apply GGOT to the simulation dataset and six disease datasets with varying disease progression rates to substantiate its effectiveness. Compared to existing methods, our proposed GGOT exhibits superior performance in detecting critical transitions.
Similar content being viewed by others

Introduction
Disease progression is a dynamic process regulated by numerous genes, involving transitions across different pathological stages, which offers critical opportunities for early diagnosis and intervention1,2. Extensive evidence suggests that sudden and catastrophic deterioration, known as critical transitions, occur during progression, with the tipping point marking the threshold for an abrupt and often irreversible shift from stability to disease3,4, and some certain key genes or proteins play a crucial role in the critical transitions. This phenomenon has been observed in diverse conditions, including epileptic seizures5,6, asthma attacks7, and cancer progression8. Prior to reaching the critical state, disease progression is often gradual, and appropriate interventions can restore the system to its normal state9,10. Identifying these transitions and key molecules is vital for understanding disease mechanisms and developing targeted therapies, as the critical state represents a critical window for intervention before irreversible deterioration occurs11,12,13.
Detecting critical transitions remains a significant challenge. Before reaching the tipping point, patients may appear stable with subtle changes that can be obscured by biological noise, patient heterogeneity, sample imbalances, and limited aligned samples9,10,12,14. As a result, critical transitions can appear abrupt and are often hard to identify in advance. With the development of high-throughput sequencing technology, gene expression data provides an excellent and abundant source of information for investigating dynamic progression15,16. Existing approaches in detecting critical transitions have explored various strategies. The critical transition has been mathematically explored in theories4 that developed a comprehensive framework involving bifurcations, fast-slow systems, and stochastic dynamics to understand and predict critical transitions in complex systems. The methods3,11,14 explored the critical transitions in complex systems by identifying early-warning signals using critical slowing down, like changes in variance, autocorrelation in system dynamics, demonstrating their applicability across ecological, climatic, and financial systems. Dynamic network biomarker methods10,12 have enhanced transition detection by identifying the dynamic changes in network structures and molecular interactions, resulting in theoretical founded and implementable framework for detecting key molecules, however may face limitations when applied to challenging heterogeneous or acute diseases. Further works17,18,19,20,21 investigate the sample-level tipping point detection based on landscape dynamic network biomarker (LDNB)17, single-sample landscape entropy (SLE)18, sample network module biomarkers (SNMB)19, sample-perturbed network entropy (SPNE)20, sample-specific causality network entropy (SCNE)21, and so on for identifying the tipping point during disease progression. These methods have provided comprehensive approaches for analyzing disease progression. However, it still lacks a systematic approach that can effectively identify global critical transition for various challenging datasets, while efficiently investigating the local trigger factors, enhancing both visualization and sample-level predictions in a principled framework.
In this work, we propose Gaussian Graphical Optimal Transport (GGOT), a framework for detecting critical transitions and identifying trigger molecules in disease progression. GGOT combines Gaussian Graphical Models (GGM)22 and Optimal Transport (OT)23. The GGM models gene interaction networks by capturing gene correlations under a Gaussian distribution24, incorporating biological priors from protein-protein interaction (PPI) network25. Optimal Transport quantifies distributional shifts between disease stages, each modeled as a Gaussian distribution using GGM, allowing for identifications of tipping points as the stage with maximal Wasserstein distance. By integrating them, GGOT detects critical tipping points while pinpointing key molecules driving the transitions, enhancing both mechanistic insights and the clinical applicability of early interventions.
Specifically, GGOT constructs disease stages by Gaussian distributions based on GGM embedded with PPI, to reduce the influence of irrelevant variables and enhance the interpretability of the model by considering the actual biomolecular associations. Then GGOT uses the Wasserstein distance to measure the distance between normal and different disease stages at the population level, which avoids inferring the inverse of the covariance directly. This distance quantifies the minimum “effort” for transitions from normal to disease stages, sensitive to gene component changes. Besides, GGOT can identify trigger molecules by decomposing the global Wasserstein distance to local Wasserstein distances, enabling the analysis of biomarkers, key pathways, and functions. Finally, GGOT predicts the probabilities of disease states for unknown samples and visualizes the disease transport process.
We validate GGOT on simulation data and six real-world datasets covering both chronic and acute diseases. Results demonstrate that GGOT can effectively detect tipping points, identify biologically relevant trigger molecules, and outperform existing methods in accuracy. For example, in lung cancer, GGOT correctly identifies the critical transition at an intermediate stage and highlights key molecules. In sepsis, We collect gene expression data from patients. GGOT uncovers trigger molecules, demonstrating its adaptability to acute disease dynamics. By addressing the limitations of existing methods and providing a comprehensive framework for disease progression analysis, GGOT offers a powerful tool for advancing precision medicine and understanding critical transitions in complex biological systems.
Results
Overview of GGOT
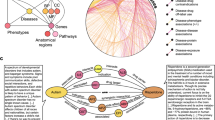
Given temporal gene expression data \({\{{S}_{i}\}}_{i = 0}^{N}\) of the specific disease with N + 1 disease stages, where \({S}_{i}=\{{g}_{i}^{\,j}\}\) denotes the set of disease samples of stage i. We assume ΩS = {0, 1, …, N} denotes all stages of the disease, with i = 0 as the normal stage and i ≠ 0 as the following abnormal stages. Dynamic progressions of diseases are characterized by multi-criticality, instability, and high complexity (Fig. 1a). As the system approaches a tipping point, it experiences significant instability resulting in increased variance and covariance among interacting components. This phenomenon, widely recognized in complex systems theory, manifests as a transient state where gene expression distributions deviate from both the healthy baseline and the subsequent disease steady state. This reflects a critical restructuring of molecular interactions, where functional relationships among genes are most perturbed at the tipping point among the various disease progression stages. Given this instability, an effective method for detecting critical transitions should move beyond simple modeling of changes in gene expression levels and instead quantify broader global network alterations and distributional shifts across the stages of disease progression. Along with this motivation, in this work, we introduce a GGOT approach to quantify the distributional distance from a healthy state to different disease states as the measure, which alleviates the challenges caused by sample imbalance and the lack of aligned samples, and it is a distribution-based framework generally robust to data noise.

a The critical transition phenomena always exhibit in disease progressions. Disease progression stages can be categorized into three states, i.e., normal, critical, and disease state, where the critical state indicates the potential for irreversible deterioration of patients. b According to the pathological characteristics, the disease samples from different stages are collected for graph modeling. The sample sizes vary typically for each stage. c The GGOT model utilizes prior knowledge from PPI networks and maximum likelihood estimation to establish Gaussian graphical distributions, characterizing the disease state for each stage. Distributions reflect dependencies between genes. d For a abnormal stage i, we aim to model it with a function Ti that maps distribution from normal stage 0 to abnormal stage i. In particular, we calculate Ti through the proposed Gaussian optimal transport due to a lack of paired measurements. e Detecting tipping points by Wasserstein distance between normal stage 0 and abnormal stage i. The critical transition appears at the stage corresponding to the maximum Wasserstein distance. f Downstream analysis. We perform downstream validation analysis of the results, including identifying trigger molecules, functional analysis, sample prediction, etc.
The proposed model GGOT has three features. First, we use the Gaussian graphical model (Fig. 1b) embedded with ___domain prior knowledge of PPI networks (Fig. 1c) to describe gene interaction networks and model data distributions in different disease stages, relieving the sample imbalance issue. Second, we model the corresponding optimal transport processes from normal to abnormal stages by the direction of disease progressions (Fig. 1d) and calculate the corresponding global Wasserstein distance to represent the minimum “effort” required to transition from normal to abnormal stages (Fig. 1e), to detect critical transitions of diseases. Last, we propose the local Wasserstein distance based on optimal transport decomposition to identify trigger molecules during critical transitions, determine biomarkers, and infer the mechanisms of disease exacerbation. We further develop downstream analytical tools to investigate the interaction relationships among molecules, predict whether an unknown sample reaches tipping points, and describe the transport processes at different stages of the disease (Fig. 1f). Our designs enable GGOT to detect critical transitions significantly and identify key molecules.
According to Gaussian graphical optimal transport, we can detect critical transitions in diseases by seeking the stage solving the maximization problem
where \({G}_{i}=G({\nu }^{{{{{\mathcal{G}}}}}_{0}},{\nu }^{{{{{\mathcal{G}}}}}_{i}}),\,{{\mbox{for}}}\,i=1,2,\ldots ,N,\) is the Global Wasserstein Distance (GWD) score from stage 0 to i determined by Eq. (9). Ti is the corresponding optimal transport map, describing the transition process. The GWD score is sensitive to compositional differences that induce global changes in the graph structure. We further decompose the GWD score Gi into the Local Wasserstein Distance (LWD) score Li of each specific gene by Eq. (11), where Li(j) measures the interaction difference of gene j under global effects. The difference in LWD scores reflects not only the changes in interactions associated with gene j but also the global structural significance of gene j (Supplementary Fig. 1). This characteristic helps to identify regulatory genes in key pathways, which is a significant advantage of our method.
Indeed, GGOT quantifies the instability of the disease network, characterized by changes in gene interactions, across different stages using the Wasserstein distance metric (Eq. (9)). In our model, we focus on variations in correlations of gene interaction patterns rather than differences in gene mean expression. The covariance element at each disease stage encodes the relational structure between genes, serving as a descriptor of the gene network. The Wasserstein distance captures the global shift between disease stage distributions relying on the covariance matrix, and measures the structural changes in gene networks. Consequently, it provides a robust measure of alterations in gene relationships during disease progression. These changes effectively reflect the instability of disease states, intensifying as the system approaches the tipping point.
To assess the effectiveness of the proposed method GGOT, we use a simulation dataset and six real-world disease datasets. The simulation dataset is generated using gene regulatory networks. The complex disease datasets (Supplementary Fig. 2) are from GEO database and TCGA database, encompassing diseases with varying rates of progression and levels of severity (Supplementary Table 4). Dimensionality reduction methods can not capture the state transition properties (Supplementary Fig. 3). The detailed information of the datasets, such as description, pathologic stages, and sample size are provided in Supplementary Section C. For each dataset, GGOT detects disease critical transitions first and quantifies the transport distances from normal to abnormal stages. Secondly, GGOT identifies trigger molecules at tipping points and performs functional analysis and survival analysis of identified molecules to illustrate regulatory mechanisms and biomarkers. GGOT then predicts the stage distributions of the unknown sample, and determines whether the sample reaches the tipping point. Finally, GGOT models the transport processes from normal to abnormal stages of diseases by optimal transport mapping and dimensionality reduction methods, such as PCA26 and T-SNE27. The results visualize the global transport processes of stages distributions and the molecule transport processes in disease progressions.
GGOT for simulation data validation
To validate GGOT, we employ a gene regulatory network in the form of Michaelis-Menten or Hill with sixteen nodes to simulate the disease progression18. Regulatory networks employing Michaelis-Menten or Hill bifurcation are frequently utilized to depict the gene regulatory activities in biological systems28,29. These networks are instrumental in detecting critical transitions. The equation (Fig. 2a) employed in our study is detailed in Supplementary Section D. The critical signal of the network is controlled by parameter q, with q = 0 as the bifurcation marking the tipping point. Nodes 1–7 are trigger molecules, directly regulated by parameter q, while the remaining nodes are irrelevant molecules, independent of parameter q. Based on the variation of the parameter q from − 0.3 to 0.2, the simulation dataset is generated to show the effectiveness of detection using GGOT when the system approaches the tipping point.

a The numerical simulation is conducted based on the graph with 16 nodes. The graph designed by the gene regulatory network reflects the relationships between nodes. b Data summary of nodes across varying conditions regulated by parameter q. The parameter q is the governing factor for the regulatory network in Supplementary Equation (S1). The left is the evolution of node 1 for different parameters q, and the right is the evolution of different nodes for q = − 0.3. c The curve of GWD score. The sudden increase in GWD scores heralds the coming of critical transition q = 0. d The landscape of LWD score. LWD scores reflect changes in single nodes within the system. The LWD scores of certain nodes, termed trigger molecules, exhibit a sudden increase as they approach the tipping point. e The changes of joint Gaussian distribution in nodes 2 and 15 between the normal stage (q = − 1) and other stages (−0.3 ≤ q ≤ 0.2). Optimal transport can accurately calculate the changes in distributions of molecular synergy. f The comparison of the distribution progression between trigger molecules and other molecules at different q. Trigger molecules are more unstable than other molecules when q = 0.
In Fig. 2b, we demonstrate changes in the value of node 1 under different parameter q settings and variations in different nodes under the parameter q = − 0.3. It shows that parameter q controls the evolution of node values. The GWD score is displayed in Fig. 2c. It can be seen that a sudden increase of GWD scores near the bifurcation q = 0 indicates the upcoming tipping point. When away from the tipping point, the GWD scores maintain a low level. To exhibit the distinct dynamics of each node in the system and identify trigger molecules during progression, we present the dynamics landscape of LWD scores in Fig. 2d. When the system is distant from the tipping point, all LWD scores are smooth and at a low level. As the system approaches the tipping point q = 0, the LWD scores of certain nodes (1, 2, …, 7) increase drastically, called trigger molecules, while the others remain low. It proves that GGOT can identify trigger molecules, and corresponding results are consistent with the nodes regulated by q. The distribution transport processes from normal to abnormal stages are illustrated in Fig. 2e. The system distribution gradually diverges and fluctuates more as it approaches the tipping point, and then returns to normal level as it moves away from the tipping point. We show the changes in marginal distributions of trigger molecules and other molecules in Fig. 2f. It indicates that the variance of trigger molecules increases dramatically approaching the tipping point. Through the simulation experiment, it is demonstrated that GGOT is reliable and accurate in detecting critical transitions and identifying trigger molecules.
GGOT detects critical transitions in disease progression, uncovering the occurrence of irreversible changes
We apply GGOT to detect critical transitions for real-world datasets with varying progression rates (Supplementary Table 4). GGOT can identify patterns of the alterations in gene relationships during disease progression, allowing for early detection of tipping points. We evaluate the effectiveness of GGOT for detecting tipping points in chronic and acute progressive diseases. The results of GSE48452, GSE2565, LUAD, and XJTUSepsis are shown in Fig. 3a–d, and the results of COAD, GSE154918 are shown in Supplementary Figs. 4a and 5a, respectively.

The curve of GWD scores in (a) GSE48452, (b) GSE2565, (c) LUAD, and (d) XJTUSepsis to detect critical transitions. GWD scores directly reflect critical changes in gene networks. The sharp increase in GWD scores suggests the upcoming tipping point and the critical transition may occur. The stage closest to the tipping point is called the pre-transition stage. Patients in the pre-transition stages are very susceptible to deterioration. e The significant difference in patient survival time before and after critical transitions (CT). Survival analysis for critical transitions in (f) LUAD and (g) XJTUSepsis is performed to demonstrate the validity of these critical transitions. The survival time of patients before critical transitions is significantly longer than the time after critical transitions (p ≤ 0.0001).
Non-alcoholic fatty liver disease (NAFLD) is a chronic progressive non-critical disease that usually begins with simple fatty liver (accumulation of fat in the liver) and may progress to non-alcoholic steatohepatitis (NASH, hepatocyte destruction, inflammation generation). GSE48452 consists of four stages, i.e., control, healthy obese, steatosis, and NASH, to analyze NAFLD. GGOT detects the tipping point of GSE48452 as the stage 2 “steatosis” (Fig. 3a). Before stage 2, liver damage is reversible, and normal liver function can be restored if measures are taken. But after stage 2, the sudden decrease in lipid correlations between blood and liver indicates the occurrence of an irreversible change30,31.
GSE2565 is employed to investigate the mechanism of lung injury, comprising ten stages determined by the time of exposure to phosgene, i.e., control, 0h, 0.5h, 1h, 4h, 8h, 12h, 24h, 48h, 72h. Lung injury is an acute progressive critical disease. GWD scores of GSE2565 demonstrate a significant increase from stage 4 to stage 5, confirming that critical transition of acute lung injury occurs at stage 5 (Fig. 3b). The disease undergoes sudden deterioration after stage 5, with 50–60% of the mice succumbing in stage 6, indicating the presence of irreversible changes beyond the tipping point. This phenomenon is consistent with the results of detection32.
Lung adenocarcinoma (LUAD) and colon adenocarcinoma (COAD) are chronic progressive non-critical diseases, with corresponding pathologic stages. The tipping point of LUAD is detected in stage IIIB, signaling the critical transition into the cancer metastasis stage (stage IV) of lung adenocarcinoma (Fig. 3c). The tumor cells invade distant tissues of other organs at stage IV, usually called advanced or metastatic cancer. After stage IIIB, the disease state deteriorates in patients, and the survival rate of patients is reduced. Critical transitions can significantly differentiate the disease states of patients (Fig. 3e). We evaluate a survival analysis of obtained patients with LUAD and compare survival curves for samples before and after the tipping point. The survival time of patients before stage IIIB is significantly longer than patients after stage IIIB (Fig. 3f). The detection signifies that GGOT serves as an early warning for cancer metastasis, and the result of COAD is similar (Supplementary Fig. 4a).
Sepsis is an acute progressive critical disease, and the previous methods can not capture progression transitions. We collect gene expression data of sepsis from the First Affiliated Hospital of Xi’an Jiaotong University, named XJTUSepsis, and group the patients based on SOFA scores33 into eight stages. GGOT effectively identifies the critical transition of sepsis at stage 5 (Fig. 3d). The SOFA score interval corresponding to stage 5 is [12, 14] (Supplementary Table 5), after which sepsis severely worsens, accompanied by the impact or failure of multiple organs. The patients may rapidly transition from a stable, critically diseased state to an extremely dangerous state. The corresponding survival analysis also demonstrates the accuracy of the sepsis tipping point (Fig. 3g). GSE154918 is another data about sepsis, and the GWD score curve is shown in Supplementary Fig. 5a. All results demonstrate that GGOT can recognize the warning signals at the early stage of the disease state transition well, which can help doctors intervene early to achieve the role of delaying disease progressions.
GGOT identifies trigger molecules to uncover the disease progression mechanisms
GGOT allows us to isolate the role of the single gene, named the LWD score, by decomposing the GWD score. The LWD scores for different diseases are illustrated in Fig. 4a–d, Supplementary Figs. 4b and 5b. It is observed that the LWD scores of specific molecules increase dramatically near tipping points, while the LWD scores of the others remain low throughout whole disease progressions. The landscapes of LWD scores in chronic diseases exhibit slower changes compared to those in acute diseases. The molecules with high LWD scores at the tipping point stage are the graph components that induce global structural changes, called the trigger molecules. These trigger molecules are more likely to play a key role in disease progression.

The landscape of smoothed LWD scores in (a) GSE48452, (b) GSE2565, (c) LUAD, and (d) XJTUSepsis to identify trigger molecules. LWD scores increase dramatically approaching the tipping point. The changes in LWD scores are mainly focused on certain molecules, called trigger molecules. The trigger molecules regulate network changes significantly and may play a greater role in disease progression. These molecules can be identified using LWD scores proposed by GGOT. We mark the trigger molecules near the critical point with boxes. Note that we illustrate interaction relationship changes of 30 trigger molecules in four disease datasets: (e) GSE48452, (f) GSE2565, (g) LUAD, and (h) XJTUSepsis. The size and color of nodes correspond to the respective scaled gene expression level, and the color of edges indicates the scaled Pearson Correlation Coefficient (PCC). As the sample approaches the tipping point, the associations between the trigger molecules gradually increase. The blue boxes are used to emphasize the interactions at tipping points.
We identify top 200 trigger molecules for different types of diseases in Supplementary Table 1. In addition, we show changes in the interactions of trigger molecules during disease progression (Fig. 4e–h, Supplementary Figs. 4c and 5c). In Fig. 4e–h, we illustrate interaction relationship changes of 30 trigger molecules using PCC in four disease datasets. The interactions between trigger molecules increase as they approach tipping points, reflecting strong correlations among these trigger molecules. To further validate the rationality of the selected trigger molecules, we perform functional and survival analysis for trigger molecules of each dataset (Fig. 5). Functional analysis can substantiate molecular feasibility and elucidate the underlying mechanisms of diseases. Concurrently, survival analysis can provide insights into the prognostic implications of these molecular findings.

We perform GO and KEGG functional analysis for (a) GSE48452, (b) GSE2565, (c) LUAD, and (d) XJTUSepsis. Different colored edges indicate different functional relationships or biological processes, circles represent different genes, and node size indicates enrichment abundance, showing the association of genes with biological processes. Box line plots of (e) LUAD and (h) XJTUSepsis demonstrate the changes in molecular expression. The blue and yellow boxes denote normal and disease states. The survival analysis of (f) MSLN, (g) RGS20, (i) SRGN, and (j) S100A9 is performed to assess significance and understand prognosis.
GSE48452 reveals that non-alcoholic fatty liver is through biological processes of chemotaxis, leukocyte migration, etc (Fig. 5a). The molecular function of the extracellular matrix structural constituent and the cellular components of the collagen-containing extracellular matrix regulate the development of nonalcoholic steatohepatitis. The major genes constituting these processes are LAMA4, THBS1, ICAM1, the CXCL family, the CCL family, and so on, emphasizing their integral involvement in the disease’s pathogenesis.
GSE2565 suggests that lung injury is related to ameboidal-type cell migration, cytoplasmic translation, etc (Fig. 5b). Specifically, cellular components of the cytosolic ribosome and the molecular function of ubiquitin protein ligase binding influence the development of phosgene lung injury. Key genes implicated in this process include Anxa1, Calr, Hmox1, Jun, members of the Rps family, etc.
LUAD indicates that lung adenocarcinoma is through biological processes such as collagen-containing extracellular matrix, epithelial cell proliferation, gland development, muscle tissue development, etc. The molecular function of glycosaminoglycan binding regulates disease trajectory in lung adenocarcinoma. The major gene clusters are MSLN, RGS20, LAMA4, CXCL family, CCL family, etc (Fig. 5c). Notably, we purposely chose MLSN and RGS20 as biomarkers to validate prognostic effects (Fig. 5e). MSLN is a cell surface antigen associated with tumor invasion and is highly expressed in a variety of cancers, acting as a cell adhesion protein. MSLN plays an important role in lung carcinogenesis and epithelial-mesenchymal transition34. Targeting MSLN is a potential CAR-T target for many common solid tumors35. We find high expression of MSLN suggests a shortened survival time of patients (Fig. 5f). It has been shown that RGS20 enhances cell cohesion response, migration, invasion, and adhesion of cancer cells36, exhibits corresponding oncogenic potential, and is an important factor in the survival of malignant adenomas of the lung37. Survival analysis showed significantly longer survival time in patients with low RGS20 expression (Fig. 5g).
XJTUSepsis demonstrates the disease progression in sepsis patients is influenced by regulating biological processes such as chemotaxis, neutrophil activation, response to lipopolysaccharide, response to molecules of bacterial origin, and secretory. The main genes that activate these terms include SRGN, S100A9, MMP9, CXCR4, CD177, and others (Fig. 5d). SRGN and S100A9 have significant differences in distribution under different states (Fig. 5h). The mortality rate of patients with high expression of SRGN and S100A9 is significantly higher than that of the low expression group (Fig. 5i, j). The secretion of SRGN is significantly increased in LPS-activated immune cells38, and SRGN is the only intracellular protein identified to load sulfated glycosaminoglycans. SRGN acts as a core protein covalently bound to different types of glycosaminoglycans and regulates the immune status of the body during different periods of disease development39,40. S100A9 in sepsis is a calcium and zinc-binding protein, playing an important role in the regulation of inflammatory processes and immune responses41. Ding and Zhao et al. demonstrate that targeted inhibition of S100A9 can reduce inflammatory responses and lung injury in sepsis42,43.
The results of COAD and GSE154918 are illustrated in Supplementary Figs. 4d and 5d. We show more detailed analysis results in Supplementary Figs. 6 and 7. The results show GGOT can identify critical transition trigger molecules as biomarkers and clarify the dynamic regulation control of trigger molecules, even under the noise influence of complex clinical therapeutic interventions.
GGOT predicts sample stage distributions, determining potential stages of unknown samples
The Gaussian graphical model of each disease stage constructed by GGOT contributes to a better understanding of disease gene interaction based on real biomolecular association networks. This is crucial for predicting whether a single sample reaches tipping points. To make predictions on unknown patients, we construct the disease stage distribution across the whole progression using Eq. (12). The stage distribution of patients provides the most direct determination of the stage at which the patient may be located. Considering the limited sample size, we use leave-one-out cross-validation to calculate the accuracy rate of GGOT in predicting whether the patient reaches the tipping point. It is worth noting that we are not predicting the stage that the sample is at, but predicting whether the state of the sample crosses tipping points. We show the prediction results in Fig. 6a, b. The numerical results are available in Supplementary Table 3. In all datasets except COAD and GSE154918, the sample prediction accuracy surpasses 0.85, and the F1 score exceeds 0.9, demonstrating the advance of the proposed GGOT.

a The unknown sample prediction accuracies of diseases with various progression rates. b The F1 score of different disease prediction results. The results are evaluated from an unbalanced perspective. We further show the predictions of disease stage distribution for a single sample in (c) GSE48452, (d) GSE2565, (e) LUAD, and (f) XJTUSepsis. We denote the tipping point boundary by dashed lines. The left half is the prediction of samples before critical transitions using blue curves and the right half is the predictions after critical transitions using red curves. Different curves indicate the potential distribution of different samples, with stars representing prediction results. GGOT effectively recognizes unknown samples that are approaching the tipping point. The stage distribution of the sample is directly related to the corresponding gene expression.
We further illustrate the corresponding stage distributions of samples before and after critical transitions to analyze patient states. (Fig. 6c–f, Supplementary Figs. 4e and 5e). It is found that the prediction outcomes for non-critical disease samples are better than those for critical disease, and samples after the tipping point are prone to be mistakenly predicted as not having reached the critical state, particularly in LUAD. Critical diseases always exhibit greater complexity and the corresponding gene networks are more similar among different stages, which results in erroneous judgments by GGOT. However, GGOT demonstrates the ability for individualized diagnosis by determining if patients are experiencing irreversible transitions, aiding in personalized diagnosis and treatment.
GGOT visualizes stage transport processes, assessing disease progression directions
GGOT offers a different perspective for measuring the state of disease progression, capable of accurately reflecting the phenomenon of disease critical transitions. The transport map T from normal to abnormal stage learned by GGOT allows us to describe the transport process of the disease progression by using Gaussian graphical distribution. The transport process of the disease is indispensable for understanding the disease progression trajectory and irreversible changes. Here, we utilize principal component analysis (PCA)26 to demonstrate the main transformation processes in the stage distribution of different diseases (Fig. 7). The change in disease distribution is directly correlated with the GWD score. The distribution is more concentrated away from tipping points, but gradually disperses as it approaches the tipping point with the increasing GWD scores. It is demonstrated the distribution transport process of GSE48452 in Fig. 7a, and the stage state changes dramatically near tipping points. The transport processes of GSE2565 show that the distribution of stage 5 is the farthest from the normal stage (Fig. 7b), revealing that the system is on the verge of collapse. The state of disease becomes worse at the next stage 6. The state of LUAD is relatively stable before the critical transition, and difficult to diagnose. After crossing the critical transition, it deteriorates significantly with distribution changing rapidly (Fig. 7c). Sepsis progresses rapidly, making it difficult to capture the changes in stages, while GGOT can accurately describe the distribution changes in sepsis and detect that stage 5 is the tipping point (Fig. 7d). We also display the results of COAD and GSE154918 in Supplementary Figs. 4f and 5f. The more detailed results across the whole stages are shown in Supplementary Figs. 8 and 9. Meanwhile, we show distributions of trigger molecules in different stages to illustrate the regulatory changes of individual molecules in disease progression (Supplementary Figs. 10 and 11). Combining the transport processes of stage and distribution changes of trigger molecules, we find that the magnitude of state transitions in acute progressive disease is higher than the magnitude in chronic progressive diseases, which is consistent with disease progression rates. Different diseases exhibit diverse patterns of transitions during progression processes, yet GGOT can capture the alterations and detect tipping points in diseases effectively.

The visualizations of the optimal transport map from normal to different abnormal stages using PCA embeddings, (a) GSE48452, (b) GSE2565, (c) LUAD, and (d) XJTUSepsis. The blue curves denote the normal stage, and the orange curves denote the abnormal stages. We focus on showcasing the changes near the tipping point. The Gaussian distribution changes little when away from tipping points, but the distribution changes dramatically near tipping points. These phenomena indicate the instability of the critical state and the suddenness of the deterioration.
Comparison of GGOT with state-of-the-art methods
We compare GGOT with six existing methods17,18,19,20,21 for detecting critical transitions. All methods use the same experimental setup and data inputs to ensure fairness (Supplementary Section E.1). We show the critical transition detection results of six datasets in Supplementary Table 2 and the quantitative comparison results in Table 1. The method of “Variance” as a baseline is implemented by extending the approach in Scheffer et al.11, by defining the variance-based metric as \({{{\rm{Var}}}}({S}_{i})=\frac{1}{d}{\sum }_{j = 1}^{d}\left({{{\rm{Var}}}}({g}\,_{i}^{j})-{{{\rm{Var}}}}({g}\,_{0}^{j})\right)\), where Si is the i-th stage of the disease, \({g}_{i}^{j}\) denotes expression values of the j-th gene at i-th stage. This method successfully detects tipping points in LUAD, COAD, and GSE154918, but fails in other datasets, as indicated by N/A, highlighting its limitations in complex disease progression.
For other sample-level methods in Supplementary Table 2, the detection results of GGOT in GSE48452, LUAD, COAD, and XJTUSepsis are consistent with other methods. In GSE2565, the results of GGOT are the same as these results of majority of methods, except LDNB. It should be emphasized that except ”Variance" only GGOT detects the tipping point in GSE154918, which demonstrates the effectiveness of GGOT in acute progressive critical disease. Some methods fail to detect critical transitions. The comparison is measured in three aspects: specific expression (CSI), certainty (SEI), and statistical difference (p-value) for critical transitions in Table 1. The results of LUAD, COAD, XJTUSepsis, and GSE48452 demonstrate better values of CSI, SEI, and p-value in uncovering critical transitions during disease progression, particularly in the cases of tumors and sepsis, as shown in Supplementary Figs. 12 and 13. The superior performance of GGOT is related to its characterization of the Wasserstein distance, which is based on modeling distributional changes. Moreover, GGOT can identify structural differences more effectively when analyzing disease progression (Supplementary Fig. 1). These results demonstrate that GGOT can reliably detect critical transitions in diseases with varying progression rates compared to existing methods.
Moreover, we conduct ablation study comparing GGOT with and without PPI network in Supplementary Table 9. GGOT with PPI achieves higher CSI scores, reflecting better identification of critical transitions. The SEI (Shannon Entropy Index) is consistently lower with PPI integration, indicating higher certainty in the detected tipping points. The p-values for GGOT with PPI are smaller in most cases except COAD, further validating the effectiveness of integrating PPI network.
Discussion
In this work, we propose Gaussian Graphical Optimal Transport (GGOT), a framework to model disease progression states from unpaired and unbalanced patients. By adequately modeling the nature of the problem through the lens of Gaussian graphical optimal transport, GGOT determines when disease states reach tipping points, identifies trigger molecules in critical transitions, predicts the sample stage distribution, and subsequently assists in a better understanding of distribution transport processes and disease functioning mechanisms. GGOT measures the disease progression instability by introducing an analytic global Wasserstein distance as the early warning signal. Previous methods rely on statistical characteristics, and can only detect tipping points without describing the transport process of disease progression. We model disease progression by using optimal transport (OT) in terms of changing gene network dynamics. This is the OT-based application for disease tipping point detection. GGOT combines data information and knowledge of biomolecular association networks to shape the true progression of the disease and understand the distribution of gene expression data for diseases at different stages. Distribution-based modeling makes GGOT more robust and stable, as evidenced by the strong performance of GGOT compared with existing methods. GGOT consistently performs well on various problems and different noisy datasets without the need for parameter tuning.
GGOT can detect critical transitions of different types of diseases without making strong assumptions about disease progressions. GGOT embeds protein interaction networks based on the data to enhance information characterization and remove noise. The GWD score as the early warning signal is more sensitive to structural components in the graph that cause global changes, which makes it a better measure of key differences. We confirm this advantage through experiments on diseases with different rates of progression (Fig. 3) and comparison with existing methods (Table 1). In particular, it performs well in high-noise acute sepsis, demonstrating its effectiveness in acute progressive diseases.
GGOT identifies trigger molecules based on LWD scores, which quantify the contribution of each gene to network changes. We depict the landscape of the LWD score across disease stages, observing that certain genes, identified as trigger molecules, exhibit a sharp increase in LWD scores near the tipping point. Conversely, the LWD scores of other genes consistently remain low, aligning with established findings regarding trigger molecules (Fig. 4a–d). Besides, the correlation between trigger molecules grows when closer to the tipping point (Fig. 4e–h). The results of functional analysis and survival analysis further validate trigger molecule significance, whose enriched gene pathways reveal key factors in the progression of the corresponding diseases, while the survival analysis helps to discover biomarkers and predict the survival of patients (Fig. 5).
We further analyze the probabilistic distribution over different stages for a single unknown sample, which allows us to predict which stage the sample is most likely to be in and to determine the severity of the patient’s illness. GGOT is highly accurate in prediction as shown in Fig. 6, which is significant to personal healthcare. We also describe the disease progression process by analyzing the optimal transport map (Fig. 7). The most significant transition in stage distribution occurs when the disease state progressively approaches tipping points. The system is at the limit of change and will undergo a critical transition. We also observe that the distribution changes are associated with the rate of disease progression. Acute progressive diseases are typically characterized by more rapid changes in distribution.
Disease progression is a complex and nonlinear physiologic process. GGOT provides a generalized framework for federating data and prior knowledge, and we can incorporate any relevant biomolecular association networks in addition to PPI networks to improve the real-world significance of the model. In a word, the use of GGOT to detect tipping points in disease provides a way for future work, including its use to improve understanding of disease progression and molecular regulatory mechanisms. GGOT makes contributions to the pathological analysis of diseases and individualized precision healthcare. Although GGOT is effective in detecting tipping points of disease progression, we have not adequately modeled the continuous process of disease change but rather treated it discretely, and not considered the effects of mutations and subtypes as well, which is an area that can be improved in the future.
Methods
The details of the theoretical background and related work can be found in Supplementary Section A.
Gaussian graphical model
Gaussian graphical models22 are probabilistic graphical models, which can represent the conditional dependencies between variables through Gaussian distributions44, e.g., the genetic association networks24 for modeling relations or dependencies of genetic factors. The Gaussian graphical model is constituted by a graph structure coupled with a Gaussian distribution.
The random vector \(X\in {{\mathbb{R}}}^{d}\) follows the multivariate Gaussian distribution \({{{\mathcal{N}}}}(\mu ,\Sigma )\) with mean vector \(\mu \in {{\mathbb{R}}}^{d}\) and covariance matrix \(\Sigma \in {{\mathbb{R}}}^{d\times d}\), where Σ is a symmetric positive semi-definite matrix. The corresponding density function is
where \(X\in {{\mathbb{R}}}^{d}\), and d denotes the number of variables in X. In addition, we denote a graph by \({{{\mathcal{G}}}}=(V,E)\) with V representing d variables in X and E denoting the set of edges among these variables, and each edge indicates the dependency between two variables.
The Gaussian graphical model is based on the multivariate Gaussian distribution44, but utilizes the graph to depict the dependency among variables in the multivariate Gaussian distribution of Eq. (1). A random vector \(X\in {{\mathbb{R}}}^{d}\) is said to satisfy the Gaussian graphical model with graph \({{{\mathcal{G}}}}\), if X has a multivariate Gaussian distribution \({{{\mathcal{N}}}}(\mu ,\Sigma )\) with Σ−1(i, j) = 0 for all (i, j) ∉ E22. Obviously, graph \({{{\mathcal{G}}}}\) describes the sparsity pattern of the precision matrix Σ−1. It shows that conditional independence relations in the Gaussian graphical model correspond to the missing edges in \({{{\mathcal{G}}}}\).
Optimal transport
Optimal transport is a theory about distribution transport, which plays a key role in transcriptomic data analysis45,46. It introduces a mathematically well-characterized distance metric, i.e., Wasserstein distance, between distributions as well as provides a geometry-based approach to realize couplings between two probability distributions23. This distance measure can be used to analyze the stability of the system state, which is instructive for critical transition warnings. Let ν1 and ν2 be two measures in \({{\mathbb{R}}}^{d}\), the Wasserstein distance between ν1 and ν223 based on optimal transport is defined as
where T is the optimal transport map corresponding to the smallest “cost” on a metric space \({{{\mathcal{X}}}}\), and T♯ν1 denotes the push-forward operation from ν1 to ν2. This formulation is non-convex and challenging to solve. However, when ν1 and ν2 are Gaussian distributions with zeros mean and Σ1 and Σ2 as covariances, i.e.,
the 2-Wasserstein distance can be written explicitly in terms of covariance matrices as
and the optimal transportation map is \(T(x)={\Sigma }_{1}^{-1/2}{({\Sigma }_{1}^{1/2}{\Sigma }_{2}{\Sigma }_{1}^{1/2})}^{1/2}\!\!{\Sigma }_{1}^{-1/2}(x)\).
The Wasserstein distance captures the distributional changes of the measures, and it can model the changes in structural information of the underlying graph represented by the covariance matrix. It is more sensitive to graphical differences that lead to global changes, rather than the differences that have little influence on the graph changes47. It is known that the gene regulatory network undergoes global changes before irreversible deterioration of the disease occurs48. As a result, for the gene regulatory network of a disease, this capability enables the discovery of critical transitions during the progression of the disease. The Wasserstein distance can effectively identify differences in network components by comparing the disease network to a normal network, which can be used to detect the critical transitions and identify the trigger molecules that contribute to the critical abrupt mutation causing the disease. Moreover, the optimal transport map T enables the movement of the disease stage from one gene graph to another, which is important for predicting the gene graph of the next disease stage. We can directly describe the complex progression of diseases by optimal transport map.
The Gaussian graphical optimal transport model
Recent high-throughput technologies provide a more in-depth understanding of disease progression. However, these data are often unbalanced in sample size across disease stages and lack temporal resolution and alignment. The disease samples are noisy, and can not necessarily provide all the information about disease progression in individual patients. In the following, we describe our approach, Gaussian Graphical Optimal Transport (GGOT) model, that detects disease critical transitions by measuring the difference between normal and abnormal stages using optimal transport, and each stage of the disease is modeled as Gaussian graphical distribution.
Constructing gene graph associated with PPI networks
The disease progression is regulated by gene interaction networks, with different stages of the disease being determined by different gene interaction networks. So we characterize the gene interaction network as a graph to depict the disease stage. The variables V, d, and E respectively denote the set of genes, the number of genes, and the gene interactions in our problems. The edge (i, j) ∈ E implies that there is a regulatory relationship between genes i and j. The graphs are described using the Gaussian graphical model. Assuming that the gene network at a particular stage of the disease follows a Gaussian distribution \({{{\mathcal{N}}}}(\mu ,\Sigma )\), we are given n independent and identically distributed observations X(1), …, X(n) from \({{{\mathcal{N}}}}(\mu ,\Sigma )\). The corresponding log-likelihood can be written as
The unbiased estimate of (μ, Σ) is derived by \(\hat{\mu }=\bar{X}\) and \(\hat{\Sigma }=\frac{1}{n-1}{\sum }_{i = 1}^{n}\left({X}^{(i)}-\bar{X}\right){\left({X}^{(i)}-\bar{X}\right)}^{T}\), where \(\bar{X}=\frac{1}{n}{\sum }_{i = 1}^{n}{X}^{(i)}\). The μ reflects the mean gene expression levels at the current stage, and the Σ characterizes the interactions between genes and determines the structure of the graph. Specifically, The covariance matrix element Σ(i, j) determines the global correlation between gene i and gene j, with Σ(i, j) = 0 indicating that gene i and j are globally independent. Meanwhile, the precision matrix elements Σ−1(i, j) reflect the local correlation between genes i and j, where Σ−1(i, j) = 0 signifies that genes i and j are locally independent after giving the remaining genes.
According to the dynamic changes of diseases and the validation of clinical information, we propose that the disease progresses through N + 1 stages, with ΩS = {0, 1, …, N} denoting the disease stage index set. Given a temporal gene expression dataset \({\{{S}_{i}\}}_{i = 0}^{N}\) encompassing N + 1 stages for the disease, where Si represents the set of disease samples at stage i containing ni samples, we assume that S0 corresponds to the sample set of patients in the normal stage and the subsequent stages Si for i > 0 are indicative of various abnormal stages. The size ni of stage Si varies with the index i, and \(\mathop{\sum }_{i = 0}^{N}{n}_{i}=D\). The indexes in ΩS are ordered in increasing stages indicating the different disease progression stages. Let \({g}\,_{i}^{j}\in {{\mathbb{R}}}^{d}\) denote the sample j in stage i, \({S}_{i}={\{{g}_{i}^{j}\}}_{j = 1}^{{n}_{i}}\), where d is the number of the genes. Therefore, we adopt \({g}_{i}^{j}(k)\) to represent the expression level of the gene k in the sample j at the stage i.
In our setting, the patients in different stages are not required to be aligned in identities across stages, which reduces the limitations for data. We assume the critical transition of disease is more determined by the changes in regulatory relationships between genes rather than the ones in the gene expression values. To eliminate the expression variation effect of different stages of data and better focus on the changes of edge relations of the gene network, we centralize the data according to the stages, by subtracting the averaged gene expression value for each gene in each stage. For each stage Si, i = 0, 1, …, N, we define the centralized sample data \({\tilde{g}}_{i}^{j}(k)\) of gene k as
where \({\sum }_{j = 1}^{{n}_{i}}{\tilde{g}}_{i}^{j}=0\). Based on this, we define a Gaussian graphical model \({{{{\mathcal{G}}}}}_{i}\) with d gene variables for Si. It is assumed that the graph is connected, undirected, and edge-weighted. The edges characterize the interactions between genes. The corresponding Gaussian distribution of the graph \({{{\mathcal{G}}}}\) is \({\nu }^{{{{{\mathcal{G}}}}}_{i}}={{{\mathcal{N}}}}(0,{\Sigma }_{i})\) following the formulation of unbiased estimate, where \({\tilde{\Sigma }}_{i}\) is the sample covariance as
The sample covariance \({\tilde{\Sigma }}_{i}\) describes the gene network at the current stage of Si. The element \({\tilde{\Sigma }}_{i}(j,k)\) estimates the correlation between genes j and k. The magnitude of \(| {\tilde{\Sigma }}_{i}(j,k)|\) reflects the strength of the interaction between genes i and j.
Moreover, the interaction of genes can be described by protein-protein interaction (PPI) networks, which contribute to analyzing the phenotype of the disease. While PPIs are not direct proxies for gene regulatory interactions, they provide valuable biological context by reflecting potential functional relationships between genes. Gene regulatory interactions can occur indirectly via proteins, metabolites, or other intermediates49. Proteins are products of genes, and PPIs provide useful evidence for gene regulation. PPI data have been effectively used in previous studies to enhance gene regulatory network inference50,51,52. For example, Yeger-Lotem et al. integrated PPI and protein-DNA interactions to identify regulatory circuits and composite network motifs50,51. Zuo et al. demonstrated that incorporating PPI priors in Graphical Lasso models improved the robustness and biological relevance of inferred networks53. Liu et al. successfully combined PPI data and gene expression matrices to detect critical transitions in disease progression18. Therefore, constructing suitable protein networks using genetically related genes in complex diseases enables providing rational hypotheses for experiments. We construct the corresponding disease PPI global network, denoted as P, utilizing the STRING database25. The elements of P(i, j) ∈ [0, 1] denote the interaction confidence levels of gene i and gene j by existing knowledge, with close to 1 indicating high confidence in a gene pair’s interaction. We incorporate prior knowledge from the PPI network P into the covariance matrix \({\tilde{\Sigma }}_{i}\) as soft constraints to enhance its descriptive ability for the real gene network, eliminating redundant relationships within the network as shown in Fig. 1c., and sparse results are shown in Supplementary Table 8. The covariance matrix Σi for the Gaussian distribution of the disease, based on the real biological significance, is defined as
where ⊙ is the Hadamard product of the matrix. We ensure that highly confident gene interactions are prioritized while allowing flexibility for weaker or unsupported interactions. The Σi provides a better approximation to the true distribution of the corresponding disease stage by taking into account both data information and biological prior knowledge. In brief, each gene edge will be assigned a corresponding confidence score. Gene pairs without strong PPI evidence are not excluded but are assigned lower weights. The integration of transcription regulation and protein-protein interaction data provides a more comprehensive view of biological networks, offering insights into functional relationships that may not be apparent from gene expression data alone. Indeed, as multi-omics data accumulate, integrating knowledge of large and heterogeneous data will provide us with additional biological insights54.
Detecting critical transitions via optimal transport
To find an effective strategy for analyzing differences across various disease stages, we build the optimal transport maps based on normal (i = 0) and abnormal (i ≠ 0) stages. For each stage i ∈ ΩS, we can obtain a Gaussian graphical model \({{{{\mathcal{G}}}}}_{i}\) whose distribution is \({\nu }^{{{{{\mathcal{G}}}}}_{i}}={{{\mathcal{N}}}}(0,{\Sigma }_{i})\). We interpret graphs as key elements that drive the probability distributions of genes in different stages. Instead of comparing patients’ gene expressions, we concentrate on the gene distributions in different stages, which are governed by the graphs. Meanwhile, optimal transport can find the minimum distance between normal and abnormal distributions. The disease state dissimilarity between normal graph \({{{{\mathcal{G}}}}}_{0}\) and abnormal graphs \({{{{\mathcal{G}}}}}_{i}(i\ne 0)\) is measured through the Wasserstein distance (Fig. 1d). It is defined as the Global Wasserstein Distance (GWD) score G, i.e.,
where Gi reflects the minimum “effort” required to recover from abnormal state i to normal state. The corresponding optimal transport map \({T}_{i}={\Sigma }_{0}^{-1/2}{({\Sigma }_{0}^{1/2}{\Sigma }_{i}{\Sigma }_{0}^{1/2})}^{1/2}{\Sigma }_{0}^{-1/2}\) denotes the push forward of \({\nu }^{{{{{\mathcal{G}}}}}_{0}}\) to \({\nu }^{{{{{\mathcal{G}}}}}_{i}}\). During implementation, we regularize the covariance matrix Σ0 and Σi by replacing negative eigenvalues with zero and adding them respectively by a scaled identity matrix (i.e., \(\Sigma =V\max (\Lambda ,0){V}^{T}+\lambda I\) with λ = 10−20) to ensure the strictly positive definiteness of them. V, Λ represent the eigenvectors, and diagonal eigenvalue matrix of the covariance matrix, and \(\max (\Lambda ,0)\) outputs the diagonal matrix with negative eigenvalues replaced by zero. The GWD score captures the structural information of the entire graph under comparison, and it is highly sensitive to differences resulting in global changes in comparison to directly comparing covariance. This allows it to precisely analyze the most significant differences in networks across different disease stages which is important for detecting the tipping point. Note that while Gaussian graphical models (GGMs) typically rely on the precision matrix Σ−1 to model the local conditional independencies between variables, the Wasserstein distance in our model, as defined in Eq. (9), does not depend on the precision matrix.
The framework described above allows us to establish maps Ti from normal (i = 0) to abnormal stages (i ≠ 0). The corresponding Gi reflects differences during stage changes, which can serve as an early warning signal for disease-critical transition. As the disease state approaches the tipping point, the dissimilarity of the disease network increases, i.e., the Gi enlarges. The internal stability of the system deteriorates at the juncture. When far from the tipping point, the dissimilarity of the disease network diminishes. Hence, we detect the tipping point I ∈ ΩS of the complex disease as
where the time point I reveals the time at which the disease reaches the critical transition point, during which the gene interaction network of the disease exhibits the maximum difference compared to the normal stage (Fig. 1e). The variations in graph components, such as the increased gene variance and the increased correlation between genes, lead to global changes in the gene network. To further validate the effectiveness of our method, we perform survival analysis.
Identifying trigger molecules in transitions
Gaussian graphical model embedded with PPI network enables the gene graph \({{{{\mathcal{G}}}}}_{i}\) to describe the gene interactions of the disease. However, Wasserstein distance only reflects the whole difference of complex interactions at different disease stages. Identifying the key regulatory genes during the critical transition period of disease occurrence is more crucial. Indeed, we can decompose the Wasserstein distance into gene-based Local Wasserstein Distances (LWD) score L by considering the complex interaction of individual genes with other genes.
The formulation of Wasserstein distance in Eq. (9) is mainly carried out by the covariance, which is related to the corresponding graph structure. The LWD score of stage i is defined as
where \({L}_{i}\in {{\mathbb{R}}}^{d}\), indicating the whole of the interaction of each single gene. We let \({\Sigma }_{0,i}=2\sqrt{{\Sigma }_{0}^{1/2}{\Sigma }_{i}{\Sigma }_{0}^{1/2}}\), where \({\Sigma }_{0,i}\in {{\mathbb{R}}}^{d\times d}\) is a symmetric positive definite square matrix. Supposing that σ0(j), σi(j), σ0,i(j) represent the j-th diagonal element in Σ0, Σi, Σ0,i respectively, we rewrite the LWD score of gene j at stage i as Li(j) = σ0(j) + σi(j) − σ0,i(j), where Li(j) is the j-th element of Li. The GWD score Gi can be decomposed as the sum of Li(j), i.e., \({G}_{i}={\sum }_{j = 1}^{d}{L}_{i}(j)\), where Li(j) reflects the difference in distribution caused by the gene j.
Considering the LWD score at tipping point I, the LI(j) denotes the contribution degree of gene j to the critical transition phenomenon at the tipping points. The significance of gene j is gauged by the magnitude of \(\left\vert {L}_{I}(j)\right\vert\), serving as a criterion to identify trigger molecules. In light of this, we can effectively identify trigger molecules by screening the top C molecules with larger LWD scores that cause the critical transition of the disease. The trigger molecules are used for the downstream analysis, which aids in disease diagnosis and gene therapy.
To further validate the regulatory mechanisms of the trigger molecules, we perform the gene functional analysis. Gene functional analysis is the process of categorizing genes according to gene prior knowledge, i.e., genome annotation information. The functional analysis including gene ontology and pathway enrichment was based on GO database and KEGG database. Gene Ontology55,56 is a functional database of the computational knowledge structure of genes, including Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). GO annotation helps to understand the biological function and significance of selected expressed genes. KEGG Pathway Enrichment57, based on biological pathways, enables the identification of the biochemical metabolic pathways and signaling pathways involved in selected expressed genes.
Predicting sample distributions for early diagnosis
Determining whether a patient reaches the critical transition point of the disease is crucial for early intervention. In addition to predicting tipping points, our model is capable of forecasting the stage distribution of unknown samples by parameterizing Gaussian graphical distributions for N abnormal stages. For a sample \(s\in {{\mathbb{R}}}^{d}\) with unknown stage, we define the probability pi of this sample being in stage i as
where \({\nu }^{{{{{\mathcal{G}}}}}_{i}}\) is the probability density function of the Gaussian graphical distribution of stage i. The probability pi reflects the relative probability of the sample potential stage i in the Euclidean space \({{\mathbb{R}}}^{d}\). Therefore, the true stage Is that sample s belongs to is predicted as
Stage Is is the period where the potential stage probability for sample s reaches its maximum. It determines the predicted stage for the sample, providing assessments of the disease severity and whether the tipping point has been reached. Due to the limited number of samples, we employ leave-one-out cross-validation for single sample stage prediction to assess the effectiveness of our method. We reserve one data point from the available dataset for prediction and train the model based on the remaining data. After repeating the experiment for D times, we compute statistics on the accuracy of the model predictions.
Visualizing disease transport processes across progressions
GGOT utilizes optimal transport to establish the distribution transformation from normal to abnormal stages of diseases. The transformation is induced by changes in gene interactions. Because the number of genes associated with the disease is extremely large, it is challenging to describe stage change trajectories in diseases under high-dimensional gene data. The optimal transport map Ti describes the transport process of the Gaussian distribution from \({\nu }^{{{{{\mathcal{G}}}}}_{0}}\) to \({\nu }^{{{{{\mathcal{G}}}}}_{i}}\). We can redefine transport maps using principal components or trigger molecules to represent the global or local distribution changes, as Ti is analytical. For the global transport process of disease, we employ principal component analysis to retain the top two components in gene samples. After data dimensionality reduction, the resulting covariance matrix is \({\Sigma }^{{\prime} }\in {{\mathbb{R}}}^{2\times 2}\). The corresponding low dimensional optimal transport map \({T}_{i}^{{\prime} }\) from \({\nu }^{{{{{\mathcal{G}}}}}_{0}}\) to \({\nu }^{{{{{\mathcal{G}}}}}_{i}}\) is represented by linear transformation. We can look across the global progression of disease progression via \({T}_{i}^{{\prime} }\). As for the trigger molecules stage changes, we consider the marginal distribution \({m}_{k,l}^{{{{{\mathcal{G}}}}}_{i}}\) of gene pair (k, l). We find \({m}_{k,l}^{{{{{\mathcal{G}}}}}_{i}}\) is still following the Gaussian distribution
The \({M}_{i}^{k,l}\in {{\mathbb{R}}}^{2}\) denotes the covariance of gene pair (k, l). The stage changes in selected trigger molecules (k, l) can be accurately characterized by \({M}_{i}^{k,l}\). We analyze changes in the relationships between several genes through the marginal distribution, unaffected by the influence of the global genes.
Model evaluation
In order to evaluate the capability of GGOT in measuring critical transition behavior, we employ three metrics to determine if there are significant differences between critical states and other states. We denote the warning signal scores of different methods as G = (G1, G2, …, GN) and the stage of tipping point as I, 2 ≤ I ≤ N − 1, where Gi represents the score at stage i. We apply the following three metrics to evaluate the performance for our research objectives. The numerical results of the model evaluations are detailed in Table 1.
-
(a)
CSI: Critical transition specific index (CSI) measures score-specific expression in the stage of critical transition58. The CSI score is defined as \(\frac{\mathop{\sum }_{i = 1}^{N}\left(1-{\eta }_{i}\right)}{N-1}\), where CSI ∈ [0, 1] is critical transition specific index score. The value \({\eta }_{i}=\frac{{G}_{i}}{\max (G)}\) is the relative level of metric score in the i-th stage, normalized by the maximal component value. The larger CSI score indicates higher confidence in the existence of the stage with higher warning signal scores than the other stages.
-
(b)
SEI: Shannon entropy index (SEI) quantifies the certainty of critical transitions based on observations59. The SEI score is defined as \(-\mathop{\sum }_{i = 1}^{N}{\eta }_{i}\log ({\eta }_{i})\), where \({\eta }_{i}=\frac{{G}_{i}}{\mathop{\sum }_{i = 1}^{N}{G}_{i}}\). The smaller SEI score suggests higher certainty of critical transition, indicating that the corresponding detection method is more confident.
-
(c)
p-value: One sample t-test statistic60 assesses whether the value GI significantly deviates from the mean of the scores (G1, …, GI−1). The corresponding statistic (p-value) from the t-distribution can assess the statistical difference between critical stages and normal stages. We compute the p-value according to the experimental setup of Zhong et al.21. The smaller p-value indicates a more significant critical transition.
Datasets and preprocessing
We apply the GGOT method to six time-course or stage-course datasets, i.e., the lung adenocarcinoma (LUAD), the colon adenocarcinoma (COAD) from TCGA database, the non-alcoholic fatty liver (GSE48452), the lung injury (GSE2565), the sepsis (GSE154918) from GEO database, and our collected dataset of sepsis patients from the First Affiliated Hospital of Xi’an Jiaotong University. For all real-world datasets, we discard the probes without corresponding NCBI Entrez gene symbols. Meanwhile, for each gene symbol mapped by multiple probes, the maximum or average value is employed as the gene expression. The procedures for selecting samples and pre-processing data are as follows.
-
Precise inclusion and exclusion criteria: We rigorously define the study population using standardized diagnostic criteria to minimize heterogeneity unrelated to the research objectives. For datasets from the GEO database, we prioritize studies that adhere to uniform experimental protocols, include comprehensive and well-defined clinical annotations, and utilize consistent diagnostic frameworks to ensure data reliability. For datasets from the TCGA database, we ensure that unrelated factors, such as gender and smoking status, are balanced across samples at each disease pathology stage (such as TCGA-LUAD, see Supplementary Table 6).
-
Normalization and preprocessing: All datasets undergo rigorous batch correction (Combat61), normalization (TPM62), and outlier removal. Additionally, non-expressed genes are filtered out to minimize technical noise and ensure the robustness of the subsequent analyses.
-
Incorporation of PPI priors: We integrate protein-protein interaction (PPI) networks into the Gaussian Graphical Model (GGM) to focus on biologically meaningful gene-gene interactions while reducing the influence of noise and irrelevant connections. This approach ensures that the resulting networks are biologically plausible and less susceptible to confounding factors.
-
Focus on population-level transitions: Our method analyzes systemic changes in gene networks during disease progression, rather than focusing on individual biomarkers. By calculating the Global Wasserstein Distance (GWD, Eq. (9)), we quantify network-level transitions to detect tipping points at a population level. This strategy reduces the impact of individual outliers and ensures the detection of global patterns.
-
Biological validation of findings: To validate the relevance of the identified tipping points, we performed functional enrichment analysis (GO/KEGG) and survival analysis for key trigger molecules. These analyses confirmed that the detected molecules and pathways are strongly associated with disease progression, supporting the robustness and biological significance of our findings.
By implementing these strategies of leveraging network-level analysis and biological priors, we ensure that our method is robust to confounding factors and capable of detecting global tipping points that represent systemic network changes during disease progression.
We then pre-screen molecules for various datasets to reduce noise, as shown in Supplementary Table 7. First, we screen suitable patient samples based on clinical information and disease progression status. The screened patients should conform to the progression direction of the disease. Second, we define two parameters r (expression rate) and h (high expression level) to select highly expressed genes in samples. The gene k is chosen if the proportion of samples with gene k exhibiting an expression level higher than h is at least r. This approach effectively eliminates genes with low or no expression in the samples, reducing the impact of irrelevant variables in the experiment. Then, the protein-protein interaction networks for Homo sapiens and Mus musculus are downloaded from STRING database25. We integrate this information into the largest global gene graph Ph for the respective species. Last, we map the genes from each dataset to the corresponding global gene graph, representing gene interactions based on prior knowledge for consequent analysis. When mapping the graph, we set the threshold u of confidence level. The edges with gene confidence levels greater than u are retained, and the genes corresponding to isolated nodes will be removed. Additionally, we conduct differential gene analysis between abnormal and normal groups to assist in selecting trigger molecules. The involved parameters are available in Supplementary Section E.1.
While our GGOT method offers a robust approach for identifying disease tipping points and corresponding triggering molecules using global/local Wasserstein distance, we acknowledge that the heterogeneity of disease is always a challenging factor. This can be alleviated by stratified analysis and data quality control, however, they are not the major focus of this work. Our major contribution lies in the development of a framework to estimate critical transition stages and assess the role of genes in disease progression. In the current approach, each stage is modeled by a Gaussian distribution without fully considering the heterogeneity. In the future work, we will explore how to more precisely model disease stage distributions considering data heterogeneity, potentially through techniques such as Gaussian mixture model. Additionally, we plan to further investigate distributional shifts and refine the identification of trigger molecules by integrating the Gaussian mixture modeling of each disease stage into the optimal transport for better modeling data heterogeneity in stages.
Inclusion and ethics statement
All authors contributed to the study design, data analysis, and manuscript preparation. This study complies with ethical guidelines, and ethical approval is obtained from the Medical Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University (Approval No. XJTU1AF2021LSK-467). All participants involved in this study provided written informed consent before their inclusion. Participants were informed about the purpose of the study, potential risks, and their right to withdraw at any time without any consequences. All ethical regulations relevant to human research participants were followed.
The XJTUSepsis dataset is collected with authorization from the First Affiliated Hospital of Xi’an Jiaotong University, ensuring compliance with patient privacy regulations. Data-sharing policies will be updated in accordance with future agreements.
Statistics and reproducibility
This study analyzes disease progression using multiple gene expression datasets. Data preprocessing steps, including batch effect correction (Combat), normalization (TPM), and feature selection, are consistently applied across all datasets to ensure comparability. Gaussian Graphical Models (GGM) are constructed with embedded prior biological knowledge from protein-protein interaction (PPI) networks. All analyses are conducted using Python (Numpy, Scipy, Torch).
The study utilizes six independent datasets from publicly available sources, including GEO and TCGA, with sample sizes detailed in Supplementary Section C. Disease samples are stratified into different pathological stages based on standardized clinical criteria. No samples are excluded unless technical artifacts are detected.
Reproducibility is assessed by validating results across multiple datasets and ensuring that tipping points and key regulatory molecules are consistently identified. The GGOT framework is tested on both simulated and real-world disease data, demonstrating its ability to detect critical transitions under different conditions. All experiments are repeated five times independently, and the results are consistent across replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The details of the simulation data are available in Supplementary Section D. Raw published data for the non-alcoholic fatty liver63, the lung injury32, the sepsis64 are available from the Gene Expression Omnibus (GEO) under accession codes GSE48452, GSE2565, GSE154918, respectively. The lung adenocarcinoma and the colon adenocarcinoma datasets are from the Cancer Genome Atlas Program (TCGA). Their original data are downloaded at the link https://portal.gdc.cancer.gov/projects/TCGA-LUAD and https://portal.gdc.cancer.gov/projects/TCGA-COAD. The sources of the above data are provided with this paper. The processing data sets for all tasks can be downloaded from https://github.com/huawenbo/GGOT. The XJTUSepsis dataset at the First Affiliated Hospital of Xi’an Jiaotong University was collected with ethical approval and in compliance with patient privacy regulations. Due to the clinical and research sensitivity of XJTUSepsis and to ensure patient confidentiality, access to this dataset requires authorization from the hospital. Please contact [email protected] to ensure that its use is in accordance with institutional policies and regulatory requirements.
Code availability
The GGOT method is written in Python and uses standard Python libraries, for detecting critical transitions and identifying trigger molecules to understand disease progression. The source code of our proposed GGOT is available at https://github.com/huawenbo/GGOT.
References
Self, W. K. & Holtzman, D. M. Emerging diagnostics and therapeutics for Alzheimer disease. Nat. Med. 29, 2187–2199 (2023).
Young, A. L. et al. Data-driven modelling of neurodegenerative disease progression: thinking outside the black box. Nat. Rev. Neurosci. 25, 111–130 (2024).
Scheffer, M. et al. Anticipating critical transitions. Science 338, 344–348 (2012).
Kuehn, C. A mathematical framework for critical transitions: Bifurcations, fast–slow systems and stochastic dynamics. Phys. D. Nonlinear Phenom. 240, 1020–1035 (2011).
Litt, B. et al. Epileptic seizures may begin hours in advance of clinical onset: a report of five patients. Neuron 30, 51–64 (2001).
McSharry, P. E., Smith, L. A. & Tarassenko, L. Prediction of epileptic seizures: are nonlinear methods relevant? Nat. Med. 9, 241–242 (2003).
Venegas, J. G. et al. Self-organized patchiness in asthma as a prelude to catastrophic shifts. Nature 434, 777–782 (2005).
Tanaka, G., Tsumoto, K., Tsuji, S. & Aihara, K. Bifurcation analysis on a hybrid systems model of intermittent hormonal therapy for prostate cancer. Phys. D: Nonlinear Phenom. 237, 2616–2627 (2008).
Achiron, A. et al. Microarray analysis identifies altered regulation of nuclear receptor family members in the pre-disease state of multiple sclerosis. Neurobiol. Dis. 38, 201–209 (2010).
Chen, L., Liu, R., Liu, Z.-P., Li, M. & Aihara, K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2, 342 (2012).
Scheffer, M. et al. Early-warning signals for critical transitions. Nature 461, 53–59 (2009).
Liu, R., Wang, X., Aihara, K. & Chen, L. Early diagnosis of complex diseases by molecular biomarkers, network biomarkers, and dynamical network biomarkers. Med. Res. Rev. 34, 455–478 (2014).
Trefois, C., Antony, P. M., Goncalves, J., Skupin, A. & Balling, R. Critical transitions in chronic disease: Transferring concepts from ecology to systems medicine. Curr. Opin. Biotechnol. 34, 48–55 (2015).
Scheffer, M. Foreseeing tipping points. Nature 467, 411–412 (2010).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Bar-Joseph, Z., Gitter, A. & Simon, I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 13, 552–564 (2012).
Liu, X. et al. Detection for disease tipping points by landscape dynamic network biomarkers. Natl. Sci. Rev. 6, 775–785 (2018).
Liu, R., Chen, P. & Chen, L. Single-sample landscape entropy reveals the imminent phase transition during disease progression. Bioinformatics 36, 1522–1532 (2020).
Zhong, J., Liu, H. & Chen, P. The single-sample network module biomarkers (sNMB) method reveals the pre-deterioration stage of disease progression. J. Mol. Cell Biol. 14, mjac052 (2022).
Zhong, J., Ding, D., Liu, J., Liu, R. & Chen, P. SPNE: Sample-perturbed network entropy for revealing critical states of complex biological systems. Brief. Bioinform. 24, bbad028 (2023).
Zhong, J. et al. Uncovering the pre-deterioration state during disease progression based on sample-specific causality network entropy (SCNE). Research 7, 0368 (2024).
Uhler, C. Gaussian graphical models: an algebraic and geometric perspective. arXiv https://doi.org/10.48550/arXiv.1707.04345 (2017).
Peyré, G. & Cuturi, M. Computational optimal transport: with applications to data science. Found. Trends® Mach. Learn. 11, 355–607 (2019).
Zhao, H. & Duan, Z.-H. Cancer genetic network inference using gaussian graphical models. Bioinform. Biol. Insights 13, 1177932219839402 (2019).
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–452 (2015).
Reich, D., Price, A. L. & Patterson, N. Principal component analysis of genetic data. Nat. Genet. 40, 491–492 (2008).
Linderman, G. C., Rachh, M., Hoskins, J. G., Steinerberger, S. & Kluger, Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 16, 243–245 (2019).
Gardner, T. S., Cantor, C. R. & Collins, J. J. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342 (2000).
O’Brien, E. L., Van Itallie, E. & Bennett, M. R. Modeling synthetic gene oscillators. Math. Biosci. 236, 1–15 (2012).
Sa, R. et al. Discovering a critical transition state from nonalcoholic hepatosteatosis to nonalcoholic steatohepatitis by lipidomics and dynamical network biomarkers. J. Mol. Cell Biol. 8, 195–206 (2016).
Pierantonelli, I. & Svegliati-Baroni, G. Nonalcoholic fatty liver disease: basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation 103, e1-e13 (2019).
Sciuto, A. M. et al. Genomic analysis of murine pulmonary tissue following carbonyl chloride inhalation. Chem. Res. Toxicol. 18, 1654–1660 (2005).
Lambden, S., Laterre, P. F., Levy, M. M. & Francois, B. The SOFA score—development, utility and challenges of accurate assessment in clinical trials. Crit. Care 23, 374 (2019).
He, X. et al. Mesothelin promotes epithelial-to-mesenchymal transition and tumorigenicity of human lung cancer and mesothelioma cells. Mol. Cancer 16, 63 (2017).
Morello, A., Sadelain, M. & Adusumilli, P. S. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. 6, 133–146 (2016).
Yang, L., Lee, M. M., Leung, M. M. & Wong, Y. H. Regulator of G protein signaling 20 enhances cancer cell aggregation, migration, invasion and adhesion. Cell. Signal. 28, 1663–1672 (2016).
Wang, Y. et al. Regulator of G protein signaling 20 correlates with long intergenic non-coding RNA (lincRNAs) harboring oncogenic potential and is markedly upregulated in hepatocellular carcinoma. Biology (Basel). 11, 1174 (2022).
Kolseth, I. B. M. et al. Serglycin is part of the secretory repertoire of LPS-activated monocytes. Immun. Inflamm. Dis. 3, 23–31 (2015).
Kolset, S. O. & Pejler, G. Serglycin: A structural and functional chameleon with wide impact on immune cells. J. immunol. (Baltim. Md, : 1950) 187, 4927–4933 (2011).
Sutton, V. R. et al. Serglycin determines secretory granule repertoire and regulates natural killer cell and cytotoxic T lymphocyte cytotoxicity. FEBS J. 283, 947–961 (2016).
Vogl, T. et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 104, 4260–4268 (2004).
Ding, Z. et al. Targeting S100A9 reduces neutrophil recruitment, inflammation and lung damage in abdominal sepsis. Int. J. Mol. Sci. 22, 12923 (2021).
Zhao, B. et al. S100A9 blockade prevents lipopolysaccharide-induced lung injury via suppressing the NLRP3 pathway. Respir. Res. 22, 45 (2021).
Bishop, C. Pattern recognition and machine learning. Springer google sch. 2, 35–42 (2006).
Zeira, R., Land, M., Strzalkowski, A. & Raphael, B. J. Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022).
Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Petric Maretic, H., El Gheche, M., Chierchia, G. & Frossard, P. GOT: An optimal transport framework for graph comparison. In Advances in Neural Information Processing Systems (eds. Wallach, H. et al.) 32 (Curran Associates, Inc., 2019).
Liu, R. et al. Identifying critical transitions and their leading biomolecular networks in complex diseases. Sci. Rep. 2, 813 (2012).
Liu, W. & Rajapakse, J. C. Fusing gene expressions and transitive protein-protein interactions for inference of gene regulatory networks. BMC Syst. Biol. 13, 37 (2019).
Yeger-Lotem, E. & Margalit, H. Detection of regulatory circuits by integrating the cellular networks of protein–protein interactions and transcription regulation. Nucleic Acids Res. 31, 6053–6061 (2003).
Yeger-Lotem, E. et al. Network motifs in integrated cellular networks of transcription–regulation and protein–protein interaction. Proc. Natl. Acad. Sci. USA 101, 5934–5939 (2004).
Zuo, Y., Cui, Y., Yu, G., Li, R. & Ressom, H. W. Incorporating prior biological knowledge for network-based differential gene expression analysis using differentially weighted graphical LASSO. BMC Bioinform. 18, 99 (2017).
Zuo, Y., Yu, G. & Ressom, H. W. Integrating prior biological knowledge and graphical LASSO for network inference. In 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 1543–1547 (IEEE, 2015).
Wang, Y.-C. & Chen, B.-S. Integrated cellular network of transcription regulations and protein-protein interactions. BMC Syst. Biol. 4, 20 (2010).
Consortium, T. G. O. et al. The gene ontology knowledgebase in 2023. Genetics 224, iyad031 (2023).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659 (2004).
Zambelli, F. et al. RNentropy: An entropy-based tool for the detection of significant variation of gene expression across multiple RNA-Seq experiments. Nucleic Acids Res. 46, e46–e46 (2018).
Gerald, B. A brief review of independent, dependent and one sample t-test. Int. J. Appl. Math. Theor. Phys. 4, 50–54 (2018).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostat. (Oxf. Engl.) 8, 118–127 (2006).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285 (2012).
Ahrens, M. et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18, 296–302 (2013).
Herwanto, V. et al. Blood transcriptome analysis of patients with uncomplicated bacterial infection and sepsis. BMC Res. Notes 14, 76 (2021).
Acknowledgements
This work was supported by National Key R & D Program 2021YFA1003002, National Natural Science Foundation of China 12125104, 62406243, 12426313. We thank the Department of SICU of the First Affiliated Hospital of Xi’an Jiaotong University, Department of Hepatobiliary Surgery and Liver Transplantation of the Second Affiliated Hospital of Xi’an Jiaotong University, and Key Laboratory of Surgical Critical Care and Life Support, Ministry of Education for their support and collecting data. We also acknowledge GEO and TCGA for providing the platforms and thank the contributors for uploading their original datasets.
Author information
Authors and Affiliations
Contributions
W.H., R.C., and J.S. conceived the method. W.H. implemented the method. W.H. and J.S. generate the numerical results. W.H., R.C. generated the experimental results. W.H., R.C., J.S., and C.L. interpreted the results. W.H., R.C., H.Y., J.Z., and J.S. generated the diagrams and wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan, Dario Ummarino.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hua, W., Cui, R., Yang, H. et al. Uncovering critical transitions and molecule mechanisms in disease progressions using Gaussian graphical optimal transport. Commun Biol 8, 575 (2025). https://doi.org/10.1038/s42003-025-07995-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07995-z
