
Abstract
Aging compromises antitumor immunity, but the underlying mechanisms remain elusive. Here, we report that aging impairs the generation of CD8+ tissue resident memory T (TRM) cells in nonlymphoid tissues in mice, thus compromising the antitumor activity of aged CD8+ T cells, which we also observed in human lung adenocarcinoma. We further identified that the apoptosis regulator BFAR was highly enriched in aged CD8+ T cells, in which BFAR suppressed cytokine-induced JAK2 signaling by activating JAK2 deubiquitination, thereby limiting downstream STAT1-mediated TRM reprogramming. Targeting BFAR either through Bfar knockout or treatment with our developed BFAR inhibitor, iBFAR2, rescued the antitumor activity of aged CD8+ T cells by restoring TRM generation in the tumor microenvironment, thus efficiently inhibiting tumor growth in aged CD8+ T cell transfer and anti-programmed cell death protein 1 (PD-1)-resistant mouse tumor models. Together, our findings establish BFAR-induced TRM restriction as a key mechanism causing aged CD8+ T cell dysfunction and highlight the translational potential of iBFAR2 in restoring antitumor activity in aged individuals or patients resistant to anti-PD-1 therapy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
118,99 € per year
only 9,92 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
All scRNA-seq data generated during this study have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE237006. Bulk RNA-seq data supporting the findings of this study have been deposited in the GEO under accession number GSE262773. Published datasets used in this study are available through the GEO under accession number GSE120575. All data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Fane, M. & Weeraratna, A. T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 (2020).
Calcinotto, A. et al. Cellular senescence: aging, cancer, and injury. Physiol. Rev. 99, 1047–1078 (2019).
Shaw, A. C., Goldstein, D. R. & Montgomery, R. R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13, 875–887 (2013).
Cai, Y. et al. The landscape of aging. Sci. China Life Sci. 65, 2354–2454 (2022).
Wang, W. et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Reina-Campos, M., Scharping, N. E. & Goldrath, A. W. CD8+ T cell metabolism in infection and cancer. Nat. Rev. Immunol. 21, 718–738 (2021).
Virassamy, B. et al. Intratumoral CD8+ T cells with a tissue-resident memory phenotype mediate local immunity and immune checkpoint responses in breast cancer. Cancer Cell 41, 585–601 (2023).
Nakajima, Y., Chamoto, K., Oura, T. & Honjo, T. Critical role of the CD44lowCD62Llow CD8+ T cell subset in restoring antitumor immunity in aged mice. Proc. Natl Acad. Sci. USA 118, e2103730118 (2021).
Dai, D. et al. Chemoradiotherapy-induced ACKR2+ tumor cells drive CD8+ T cell senescence and cervical cancer recurrence. Cell Rep. Med. 5, 101550 (2024).
Fourcade, J. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186 (2010).
Herbig, U., Jobling, W. A., Chen, B. P. C., Chen, D. J. & Sedivy, J. M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 14, 501–513 (2004).
Pereira, B. I. et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 21, 684–694 (2020).
Sakuishi, K. et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010).
Liu, X., Hoft, D. F. & Peng, G. Senescent T cells within suppressive tumor microenvironments: emerging target for tumor immunotherapy. J. Clin. Invest. 130, 1073–1083 (2020).
Liu, X. et al. Blockades of effector T cell senescence and exhaustion synergistically enhance antitumor immunity and immunotherapy. J. Immunother. Cancer 10, e005020 (2022).
Zhao, Y., Shao, Q. & Peng, G. Exhaustion and senescence: two crucial dysfunctional states of T cells in the tumor microenvironment. Cell. Mol. Immunol. 17, 27–35 (2020).
Blank, C. U. et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 19, 665–674 (2019).
Byrne, A. et al. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nat. Rev. Clin. Oncol. 17, 341–348 (2020).
Kok, L., Masopust, D. & Schumacher, T. N. The precursors of CD8+ tissue resident memory T cells: from lymphoid organs to infected tissues. Nat. Rev. Immunol. 22, 283–293 (2022).
Mueller, S. N. & Mackay, L. K. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 16, 79–89 (2016).
Schenkel, J. M. & Masopust, D. Tissue-resident memory T cells. Immunity 41, 886–897 (2014).
Amsen, D., van Gisbergen, K. P. J. M., Hombrink, P. & van Lier, R. A. W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 19, 538–546 (2018).
Okła, K., Farber, D. L. & Zou, W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 218, e20201605 (2021).
Ganesan, A.-P. et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 18, 940–950 (2017).
Jain, A., Sturmlechner, I., Weyand, C. M. & Goronzy, J. J. Heterogeneity of memory T cells in aging. Front. Immunol. 14, 1250916 (2023).
Sturmlechner, I., Jain, A., Mu, Y., Weyand, C. M. & Goronzy, J. J. T cell fate decisions during memory cell generation with aging. Semin. Immunol. 69, 101800 (2023).
Clegg, A., Young, J., Iliffe, S., Rikkert, M. O. & Rockwood, K. Frailty in elderly people. Lancet 381, 752–762 (2013).
Enamorado, M. et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 8, 16073 (2017).
Sade-Feldman, M. et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013 (2018).
Ye, J. et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 6, 1294–1311 (2014).
Wang, L. et al. FBW7 mediates senescence and pulmonary fibrosis through telomere uncapping. Cell Metab. 32, 860–877 (2020).
Liu, X. et al. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 9, 249 (2018).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 (2005).
Ivashkiv, L. B. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018).
Hunter, C. A. & Jones, S. A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 (2015).
Ross, S. H. & Cantrell, D. A. Signaling and function of interleukin-2 in T lymphocytes. Annu. Rev. Immunol. 36, 411–433 (2018).
Watford, W. T. et al. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol. Rev. 202, 139–156 (2004).
Pei, S. et al. BFAR coordinates TGFβ signaling to modulate Th9-mediated cancer immunotherapy. J. Exp. Med. 218, e20202144 (2021).
Zhang, N. & Bevan, M. J. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013).
Chen, L. et al. Sequence-based drug design as a concept in computational drug design. Nat. Commun. 14, 4217 (2023).
Du, L. et al. LINC00926 promotes progression of renal cell carcinoma via regulating miR-30a-5p/SOX4 axis and activating IFNγ–JAK2–STAT1 pathway. Cancer Lett. 578, 216463 (2023).
Li, N. et al. ARID1A loss induces polymorphonuclear myeloid-derived suppressor cell chemotaxis and promotes prostate cancer progression. Nat. Commun. 13, 7281 (2022).
Martins, F. et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol. 20, e54–e64 (2019).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Tsuji, T., Matsuzaki, J. & Odunsi, K. Tissue residency of memory CD8+ T cells matters in shaping immunogenicity of ovarian cancer. Cancer Cell 40, 452–454 (2022).
Mackay, L. K. et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016).
Zhou, M. et al. JAK/STAT signaling controls the fate of CD8+CD103+ tissue-resident memory T cell in lupus nephritis. J. Autoimmun. 109, 102424 (2020).
Philips, R. L. et al. The JAK–STAT pathway at 30: much learned, much more to do. Cell 185, 3857–3876 (2022).
Sun, Q. et al. STAT3 regulates CD8+ T cell differentiation and functions in cancer and acute infection. J. Exp. Med. 220, e20220686 (2023).
Guo, Y. et al. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 22, 746–756 (2021).
Feng, Q. et al. Lactate increases stemness of CD8+ T cells to augment anti-tumor immunity. Nat. Commun. 13, 4981 (2022).
Chen, Y. et al. BATF regulates progenitor to cytolytic effector CD8+ T cell transition during chronic viral infection. Nat. Immunol. 22, 996–1007 (2021).
Acknowledgements
This research was supported by grants from the CAS Project for Young Scientists in Basic Research (YSBR-076), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39030300), the National Natural Science Foundation of China (82425026, 82030041, 82201921, 82373072, 82273463), the National Key R&D Program of China (2018YFA0902703), programs from Shanghai Municipal Science and Technology (21140905000), the Natural Science Foundation of Hebei Province (H2022201067), and CAS Key Laboratory of Tissue Microenvironment and Tumor. We also thank NovelBio Co., Ltd. for the support with bioinformatics analysis using their Novel Brain Cloud Analysis Platform (https://www.novelbrain.com). We also appreciate the assistance from personnel at the animal and platform core facilities of the Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
S.P. and X.D. designed the study, performed the experiments, prepared the figures and wrote parts of the manuscript. R.Y., S.Z. and H.Hou. contributed to the screening of BFAR inhibitors. H.W., J.-H.S., J.H. and Y.T. contributed to the collection and analysis of human samples. S.P., R.W. and H. Huang contributed to the analysis and interpretation of scRNA-seq data. X.W., J.X., Q.Z. and J.Y. contributed to the experiments and data analysis. C.-Y.W. provided Jak2-floxed mice. W.L. contributed to the acquisition of CD8-Cre mice. Z.-Y.N., Q.L. and M.Z. supervised a specific subset of the experiments and analyses. Y.X. initiated, designed and supervised this study, prepared the figures, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y.X., M.Z., S.P., X.D., R.Y., S.Z. and H. Hou have filed a patent application regarding the application of the BFAR inhibitor for the treatment of tumors. The other authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks Vassiliki Boussiotis, Tyler Curiel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Aging specifically impaired the generation of CD8+ TRM cells in NLTs.
a-d, Flow cytometric analysis of the frequencies and absolute numbers of CD4+TRM (CD103+CD69+), CD4+/CD8+ TCM (CD62L+CD44+) and CD4+/CD8+ TEM (CD62L−CD44+) cells in the liver and lung tissue of young (5-8-week-old) and aged (18-24-month-old) mice. Data are presented as representative plots (a, c) and summary graphs (b, d) (n = 3 mice/group). e-h, Flow cytometric analysis of the frequencies and absolute numbers of CD8+ TEM (CD62L−CD44+) and CD8+ TCM (CD62L+CD44+) cells (e,f), or flow cytometric analysis of the frequencies and absolute numbers of IFNγ-producing CD8+ TRM cells (g,h) of Rag1-KO mice that received young (2-month-old) or aged (18-month-old) mice-derived splenic CD8+ T cell and then were s.c. inoculated with MB49 tumor cells at the same day (n = 5 mice/group). i,j, Experimental scheme (i) and tumor growth (j) of Rag1-KO mice that were s.c. inoculated with MB49 tumor cells, and then intratumorally injected with equal numbers of tumor-infiltrating CD8+ TRM cells (105/mice) that were isolated from young/aged tumor-bearing mice (n = 5 mice/group). Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of two (i, j) or three independent experiments (a-h).
Extended Data Fig. 2 Aging-induced decline in TRM is unrelated with cell proliferation, apoptosis and trafficking.
a, Scheme showing mixed young/aged mice-derived splenic CD8+ T cells transfer into Rag1–/– mice for further analysis. b, Flow cytometric analysis of TRM cells in CD8+ T cell from lung, liver and kidney of Rag1–/– mice (n = 4 mice/group). c,d, Flow cytometric analysis of annexin V expression and the percentage of Ki-67+ cells in CD8+ T cells of Rag1−/− mice were adoptively transferred with young or aged-derived splenic CD8+ T cell and s.c. inoculated with MB49 tumor cells (n = 5 mice/group). e, Experimental scheme of Rag1-KO mice that were s.c. inoculated with MB49 tumor cells, and then intratumor injected with mixed young/aged mice-derived splenic CD8+ T cells as indicated. f,g, Flow cytometric analysis of the percentage of TRM/TEM/TCM cells in CD8+ T cells from mice as described in E (n = 5 mice/group). h-j, Tumor growth (h), Flow cytometric analysis (i) and the corresponding statistical analysis (j) of the frequencies and absolute number of tumor infiltrating CD8+CD69+CD103+ TRM cells of young/aged mice that were s.c. inoculated with MB49 tumor cells (n = 5 mice/group). k,l, Experimental scheme (k) and tumor growth (l) of Rag1–/– mice that were transferred with young (2-month-old) and aged (18-month-old)-derived splenic CD8+ T cells, and then intratumorally injected with PBS or TRM cells (n = 6 mice/group). The transferred CD8+CD69+CD103+TRM cells were sorted directly from tumor tissue of young mice that were inoculated with MB49 tumor. Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of two (a,b,e-l) or three independent experiments (c, d).
Extended Data Fig. 3 CD8+ T cells from aged mice show decreased TRM inducibility and compromised anti-tumor immunity.
a,b, Heatmap (a) and corresponding GSEA (b) analysis showing differentiated gene expression profiles in young and aged mice-derived CD8+ T cells differentiated under TCR condition or TRM condition for 3 days. c,d, Flow cytometric analysis of the percentage of TCM/TEM cells in CD8+ T cells from spleen of young and aged mice. e,f, Flow cytometric analysis of TRM inducibility of naïve/memory CD8+T cells from spleen of young/aged mice under TRM condition for 3 days (n =3, biological replicates). g, Heat map of the expression levels of selected genes in different scRNA-seq clusters of tumor-infiltrating CD8+ T cells. h, FeaturePlot showing signature gene expression of different characteristic subpopulations of tumor-infiltrating CD8+ T cells. i,j, Flow cytometric analysis (i) and the corresponding statistical analysis (j) of IFNγ-producing CD8+ T cells, CD8+ TCM (CD62L+CD44+) and CD8+ TEM (CD62L−CD44+) cells of Rag1−/− mice that were adoptively transferred with young (2-month-old) and aged (18-month-old)-derived splenic CD8+ T cells and then s.c. inoculated with MB49 tumor cells at the same day, and then treated with α-CD103 antibody or control antibody (IgG) once every 3 days starting from day 7 (n = 6 mice/group). Bar graphs are presented as mean ± s.e.m. A tw-tailed Student’s t-test was performed for comparisons. The data are representative of two (i, j) or three independent experiments (c-f).
Extended Data Fig. 4 BFAR was highly expressed in aged/senescent CD8+ T cells.
a,b, Dot plots showing the expression levels of BFAR, Xaf1 and Usp18 in aged vs. young CD8+ T cells, and in TRM vs. non-TRM cells based on scRNA-seq data. c-e, FeaturePlot showing the expression of BFAR, XAF1 and USP18 in CD8+ T cells from tumor samples of non-responders (NR) vs. responders (R) after anti-PD-1 therapy (GSE120575). f, qPCR analysis of Bfar mRNA expression in splenic CD8+ T cells of young and aged tumor bearing mice. g, qPCR analysis of Cdkn1a, Xaf1 and Usp18 mRNA expression in tumor-infiltrating CD8+ T cells from MB49-bearing mice on early (day 10) and advanced tumor stage (day 20). h, qPCR analysis of Xaf1 and Usp18 mRNA expression in mouse CD27+ (Non-sen) and CD27− (Sen) CD8+ T cells that were derived from the coculture of mouse splenic CD8+ T cells with MB49 tumor cell supernatant in the presence of plate-bound anti-CD3 plus anti-CD28 (α-CD3/28; 1μg/ml) for 3 days. i, QPCR analysis of the mRNA expression of BFAR and Cdkn2a in mouse splenic CD8+ T cells that treated as indicated. j-n, Flow cytometric analysis of γH2AX and CD27 expression and qPCR analysis of the mRNA expression of Cdkn1a, Cdkn2a and BFAR in mouse splenic CD8+ T cells that treated with MB49 tumor cell supernatant or bleomycin or cocultured with tumor cells in the presence of plate-bound anti-CD3 plus anti-CD28 (α-CD3/28; 1μg/ml) for 3 days. Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of three independent experiments (f-n).
Extended Data Fig. 5 BFAR deficiency in CD8+ T cells from aged mice restored the aging-induced TRM decline.
a, Schematic picture of BFAR gene targeting to generate CD8+ T cells conditional knockout mice (BFARCD8-KO) by crossing Bfar-floxed mice with Cd8-Cre mice. b, qPCR analysis of BFAR mRNA expression in naive CD4+ and CD8+ T cells isolated from WT and BFARCD8-KO mice. c-f, Flow cytometric analysis of the frequencies and absolute number of the indicated lymphoid immune cells in the thymus, spleen, and peripheral lymph nodes of WT and BFARCD8-KO mice. Data are presented as representative plots and summary bar graphs. DP, double positive; DN, double negative (n = 3 mice/group). g, qPCR analysis of Ifng and Gzmb mRNA expression in tumor-infiltrating CD8+ T cells from WT and BFARCD8-KO mice that were injected s.c. with MB49 tumor. h-k, Flow cytometric analysis (h,j) and the corresponding statistical analysis (i,k) of the frequencies of CD8+CD69+CD103+ TRM cells and IFNγ-producing CD8+ T cells of Rag1−/− mice that adoptively transferred with young/aged WT and BFAR-deficient splenic CD8+ T cells as indicated and then s.c. inoculated with MB49 tumor cells at the same day, or treated with α-CD103 antibody or control antibody (IgG) as indicated once every 3 days starting from day 7 (n = 6 mice/group). Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of two (h-k) or three (a-g) independent experiments.
Extended Data Fig. 6 BFAR does not affect TCR and JAK1 downstream signaling activation.
a, Immunoblot analysis of phosphorylated and total ZAP70, LAT, AKT, FOXO1, ERK and p65 in whole-cell lysates of WT and BFAR-deficient splenic naive CD8+ T cells that were stimulated with anti-CD3 plus anti-CD28 (α-CD3/CD28; 1 μg/ml) for the indicated time points. b, Flow cytometric analysis of the phosphorylated STAT1 in mouse splenic CD27+CD8+ T cells or CD27−CD8+ T cells that were cocultured with MB49 tumor cell supernatant in the presence of plate-bound anti-CD3 plus anti-CD28 (α-CD3/28; 1 μg/ml) for 3 days. c-f, Immunoblot analysis of phosphorylated and total STAT3, STAT5 and STAT1 in whole-cell lysates of WT and BFAR-deficient mouse splenic naive CD8+ T cells that were stimulated with IL-6 (100 ng/ml) or IL-2 (50 ng/ml) or IFN-α (100 ng/ml) or IFN-β (100 ng/ml) for the indicated time points. g, STAT1 luciferase activity in HEK293T cells transfected with luciferase reporter, together with the indicated plasmids. h,i, Flow cytometric analysis and the corresponding statistical analysis of the frequencies of IFNγ-producing and Granzyme B-producing CD8+ T cells in WT and BFAR-deficient mouse splenic CD8+ T cells that were treated with DMSO or NF-κB inhibitor (Pyrrolidinedithiocarbamate ammonium) or JAK2 inhibitor (XL019) and were stimulated by plate-bound anti-CD3 plus anti-CD28 (α-CD3/28; 1 μg/ml) for 3 days. j, Immunoblot analysis of phosphorylated and total Smad2/3 in whole cell lysates of WT and BFAR-deficient mouse splenic CD8+ T cells that were stimulated with α-CD3/28 (3 μg ml−1) overnight and then stimulated with TGFβ1 (5 ng ml−1) for the indicated time points. k,l, Tumor growth (k) and Flow cytometric statistical analysis (l) of the frequencies and absolute number of CD8+CD69+CD103+ TRM cells and IFNγ-producing CD8+ T cells of Rag1−/− mice that were adoptively transferred with Tgfbr1+/+ and Tgfbr1K268R mouse splenic CD8+ T cell and then s.c. inoculated with MB49 tumor cells at the same day (n = 5 mice/group). Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of two (k,l) or three (a-j) independent experiments.
Extended Data Fig. 7 BFAR mediates JAK2 deubiquitination.
a-c, Immunoblot analysis of the interaction of BFAR with IFNγR1 or IFNγR2 or STAT1 in HEK293T cells transfected with the indicated expression vectors. d-i, Ubiquitination of JAK2 in HEK293T cells transfected with the expression vector encoding HA-BFAR, Flag-JAK2 and different types of Ub as indicated. The data are representative of three independent experiments.
Extended Data Fig. 8 BFAR restrains CD8+ T cell function through USP39.
a,b, Immunoblot analysis of the interaction of BFAR with USP7 or USP9X in HEK293T cells transfected with the indicated expression vectors. c, Immunoblot analysis of the interaction of JAK2 with USP9X in HEK293T cells transfected with the indicated expression vectors. d, Ubiquitination of JAK2 in HEK293T cells transfected with the expression vector encoding USP9X, Flag-JAK2 and HA-Ub as indicated. e,f, Ubiquitination of USP9X and USP39 in HEK293T cells transfected with the expression vector encoding Flag-USP9X, Flag-USP39, HA-BFAR and Ub as indicated. g-l, qPCR analysis of the mRNA expression of Bfar and Usp39, immunoblot of phosphorylated (p-) and total STAT1 and flow cytometric analysis of the proliferation and the frequencies of IFNγ-producing CD8+ T cells of WT and BFAR-deficient mouse splenic CD8+ T cells that were reconstituted with EV (control) or LMP-shUSP39 vector and stimulated by plate-bound anti-CD3 plus anti-CD28 (α-CD3/28; 1μg/ml) for 3 days. Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of three independent experiments.
Extended Data Fig. 9 iBFAR2 suppresses tumor growth with low cellular and tissue toxicity.
a, Flow cytometric analysis of proliferation and IFNγ-producing CD8+ T cells of mouse splenic primary CD8+ T cells that were treated with different concentration of iBFAR2 as indicated and stimulated by plate-bound anti-CD3 plus anti-CD28 (α-CD3/28; 1μg/ml) for 3 days. b, Tumor growth of WT mice that were injected s.c. with MB49 tumor cells and then treated with different concentration of iBFAR2 as indicated or vehicle once every 2 days starting from day 7 (n = 9 mice/group). c, Tumor growth of WT mice that were injected s.c. with Py8119 tumor cells (n = 7 mice/group) or B16-F10 melanoma (n = 6 control, n = 8 mice iBFAR2) and then were treated with iBFAR2 (10 mg/ml) or vehicle every 2 days starting from day 7. d, Flow cytometric analysis of tumor-infiltrating CD8+ TRM cells, IFNγ-producing CD8+ TRM cells, IFNγ-producing and TNFα-producing CD8+ T cells as indicated in MB49 tumor-bearing mice that treated with iBFAR2 (10 mg/kg) or vehicle (n = 4 mice/group). e, Immunoblot of phosphorylated (p-) and total STAT1 in MB49 tumor cells. f, The MB49 tumor cells were treated with DMSO or iBFAR2 for the indicated time points based on CCK8 assay. g, Tumor growth of Rag1−/−mice that were i.v. injected with young and aged mice derived splenic CD8+ T cells and s.c. injected with MB49 tumor cells, and then treated with vehicle (Veh) or iBFAR2 (20 mg/kg) once every 2 days starting from day 9 (n = 5 mice). h, Flow cytometric analysis of apoptosis of mouse splenic primary CD8+ T cells that were treated with different concentration of iBFAR2. i, qPCR analysis of the mRNA expression of Il1b and Tnf of different tissues from the tumor-bearing mice. Bar graphs are presented as mean ± s.e.m. A two-tailed Student’s t-test was performed for comparisons. The data are representative of two (b, c, g, i) or three independent experiments (a, d, e, f, h).
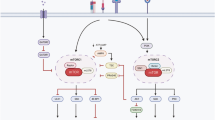
Extended Data Fig. 10 Working model of aging-induced decline of CD8+ TRM cells in compromising antitumor immunity through BFAR.
In aged CD8+ T cells, the stressed environment upregulates the expression of BFAR, which activates USP39 to mediate the deubiquitylation of JAK2. This process results in the suppression of JAK2 downstream signaling, which in turn leads to a decline in CD8+ TRM cells and consequently impaired antitumor defensive immunity. In contrast, low level of BFAR in young CD8+ T cells result in the activation of JAK2 signaling, which in turn promotes the expression of cytotoxic- and TRM-related genes. This, in turn, promotes the generation of CD8+ TRM cells and the enhancement of antitumor defensive immunity. The increased production of cytotoxic cytokine IFNγ also served to reinforce this process, thereby establishing a positive feedback loop in young CD8+ T cells. Moreover, targeting BFAR though the compound iBFAR2 specifically reinvigorated aged CD8+ T cells by restoring TRM cell subset generation, thus efficiently suppressing tumor growth in aged or PD-1-therapy-resistant individuals.
Supplementary information
Supplementary Information
Supplementary Tables 1–5 and figure.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Unprocessed western blots.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed western blots.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pei, S., Deng, X., Yang, R. et al. Age-related decline in CD8+ tissue resident memory T cells compromises antitumor immunity. Nat Aging 4, 1828–1844 (2024). https://doi.org/10.1038/s43587-024-00746-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-024-00746-5
