
Abstract
Due to the high reactivity and versatility of benzenesulfonothioates, significant advancements have been made in constructing C-S bonds. However, there are certain limitations in the synthesis of S-thiosulfonates and SS-thiosulfonates, especially when dealing with substantial steric hindrance, which poses a significant challenge. Herein, we present an innovative approach for assembling unsymmetric S-thiosulfonates and unsymmetric SS-thiosulfonates through the integration of dual copper/photoredox catalysis. Moreover, we also realized the one-pot strategy by directly using carboxylic acids as raw materials by in-situ activation of them to access S-thiosulfonates and SS-thiosulfonates without further purification and presynthesis of NHPI esters. The envisaged synthesis and utilization of these reagents are poised to pioneer an innovative pathway for fabricating a versatile spectrum of mono-, di-, and polysulfide compounds. Furthermore, they introduce a class of potent sulfenylating reagents, empowering the synthesis of intricate unsymmetrical disulfides that were previously challenging to access.
Similar content being viewed by others
Introduction
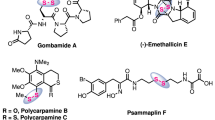
Organosulfur compounds have received considerable attention in the past decades, mainly because they are prevalent in natural products and biologically active molecules, and they are also extensively used in materials chemistry1,2,3,4,5,6. The traditional approach to access thioether compounds mainly relies on the use of thiol, disulfide and sulfinyl halide as starting materials, which suffer from strong odors, limited availability and reactivity7,8,9,10. Thus, considerable effort has been devoted to the development of new sulfurization reagents (mercaptan, disulfide, sulfinyl halide, sulfonium salt, quinone mono O, S-acetal, p-toluenesulfonyl hydrazine S-acetal, arylsulfonyl chloride, sulfite, thiosulfonates and p-toluenesulfonic acid ester etc.) as starting materials11,12,13,14,15,16. Among them, thiosulfonates were the most widely studied due to their high reactivity and biological activity17,18,19,20,21,22. For example, due to the unique properties of thiosulfonates, it can be used as filler23,24,25,26. Meanwhile, it can also be used as the key intermediate in the synthesis of cephalosporin compounds and antibiotic inhibitors (Fig. 1a)27,28,29,30. Additionally, due to their high reactivity and diversity, numerous new reactions involving thiosulfonates have been disclosed. The reactivity of thiosulfonates mainly includes four aspects: (1) As electrophilic reagents, they could react with organometallic reagents or used in metal-catalyzed cross coupling reactions (Fig. 1b, 1)21,31,32,33,34. In 2014, Ruijter et al. reported a one-step synthesis of isothioureas with isocyanates, S-methyl methanethiosulfonate, and (hetero)aromatic amines catalyzed by copper catalysis. Xu et al. also developed a copper(I)-catalyzed interrupted click reaction using S-methyl benzenesulfonothioate to obtain different 5-functionalized triazoles. (2) As radical acceptors in radical chemistry, enabling the construction of C-S bonds (Fig. 1b, 2)35,36,37,38,39,40. Ji et al. reported an effective organic photocatalytic cross coupling of 4-alkyl-1,4-dihydropyridine with thio/selenium sulfonates. (3) As 1,2-thiosulfonylating reactants (Fig. 1b, 3)41,42,43,44,45,46. Shen was the first to achieve bifunctionalization using inactivated olefins and difluoromethylthiol reagents (PhSO2SCF2H) catalyzed by silver catalyst and potassium persulfate as stoichiometric oxidant. (4) As a partner in reductive coupling reactions (Fig. 1b, 4)47,48,49, Ji et al. developed a Ni-catalyzed reductive cross-coupling of alkyl bromides with thio/selenium sulfonates to synthesize sulfides and selenides.

a Representative significant disulfanes. b The generation and application of thiosulfonates. c From sodium sulfite to thiosulfonates and trisulfide dioxides via photocatalysis (this work).
As mentioned above, the applications of thiosulfates are extensive; however, there is a scarcity of information regarding their synthetic methods as well as the synthesis of S-alkyl thiosulfonates50,51,52 and SS-alkyl thiosulfonates53,54 (Fig. 1b, top). The classic method for synthesizing S-alkyl thiosulfonates involves the sulfenylation of sodium sulfinates with disulfides in the presence of I2 or NBS. In general, S-alkyl thiosulfonates are prepared by sulfonyl chlorides with thiols catalyzed by Et3N. For SS-alkyl thiosulfonates, the traditional method was to use disulfide compounds to react with TsSK/TsSNa under the catalysis of sulfonyl chloride. In 2021, Jiang reported a photo-catalyzed synthesis of SS-alkyl thiosulfonates from sulfides and tetrasulfides. Despite significant progress made in constructing C-S bonds through thiosulfonates, there are certain limitations in the synthesis of S-thiosulfonates and SS-thiosulfonates, especially those with significant steric hindrance remain a formidable challenge in chemical community.
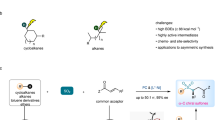
In recent years, photocatalytic decarboxylation has emerged as a well-known method to produce aliphatic radicals, especially for generating tertiary alkyl radicals55,56,57,58,59,60. N-hydroxyphthalimide (NHPI) esters have recently been widely investigated as alkyl radical precursors. They are subsequently coupled with radical acceptors or nucleophiles to achieve arylation, alkylation, halogenation, borylation or amidation reactions61,62,63,64,65. But the S-sulfonylation or SS-sulfonylation of NHPI esters has not been achieved yet. Recently, Liu reported a copper-catalyzed enantioconvergent C(sp3)-S cross-coupling using racemic secondary and tertiary alkyl halides, which also provides a new strategy for the construction of chiral thioether compounds66. Here, we reported a method for the synthesis of S-thiosulfonates and SS-thiosulfonates via the merging of photo-organocatalysis and copper-catalysis enables the decarboxylative radical S-sulfonylation or SS-sulfonylation with sodium benzenesulfonothioates at room temperature under redox-neutral conditions (Fig. 1c). This method is efficient, convenient, and especially to those substrates with large steric hindrance, and is applicable to the late-stage synthesis and modifications of bioactive molecules. Of note, the conversions can be achieved through in-situ activation of carboxylic acids without the need for additional preparation of pure N-hydroxyphthalimide (NHPI) esters with a slight decrease in yield, further demonstrating the universality and mild reaction conditions of the reaction. Moreover, the resulting S-sulfonylation or SS-sulfonylation provided a class of powerful sulfenylating reagents for the synthesis of challenging unsymmetric disulfides under mild conditions. The rapid construction of various types of thioethers, including the efficient synthesis of previously challenging unsymmetric bulky alkyl disulfides, provides a strong support for the subsequent applications of these reagents. The synthesis and application of such reagents are believed to provide a new pathway for the synthesis of various di- and polysulfide compounds, offering organic and medicinal chemists a simpler and more effective synthetic strategy.
Results and discussion
We commenced our study by using tert-butyl NHPI ester (1a) and sodium benzenesulfonothioate (2a) as the model substrates for the optimization of reaction conditions. As shown in Table 1, the desired cross-coupling product 3a was obtained in 48% yield when using 4CzIPN, a cheap and metal-free organic dye as photocatalyst, Cu(OTf)2 as catalysts, K2CO3 as additive, ethyl acetate (EA) as solvent, and under the irradiation of a 5 W blue LED lamp (entry 1, Table 1). Meanwhile, dithiol product SS-thiosulfonate 4a was also formed in 10% yield. Subsequently, we screened the copper salts, photocatalysts and solvents (entries 2-8, see Supplementary Information for details), eventually, product 3a was obtained exclusively in excellent isolated yield (entry 3, Table 1). When CF3COOH, HOP(O)(OBu)2 and other acids were used as additives, we found that the yield of product of 4a increased while 3a was almost absent (entries 9-10. See Supplementary Information for details). Solvent examinations showed that EA was the optimal one to deliver the target product 4a (entries 10–12. See Supplementary Information for details). A screening of photocatalysts, demonstrated that 4CzIPN was the most effective one, yielding trisulfide 4a in 83% yield (entries 13–15, Table 1, see Supplementary Information for details). Light, photocatalyst, and copper catalyst were all essential for the coupling reactions (see Supplementary Table 8 in Supplementary Information for details).
Substrate scopes
Upon establishing the optimized conditions (entry 3, Table 1), we firstly explored the range of substrates for NHPI esters. With tert-butyl-substituted NHPI esters, as seen in Fig. 2, we were able to generate product 3a in 68% isolated yield. Under the standard conditions, 3-, 5-, and 6-membered substituted tert-alkyl NHPI esters were thereafter able to achieve the target products (3b–3d). Similarly, for NHPI ester generated from bicyclo[2.2.2]octylcarboxylic acid, the corresponding desired product 3e was also smoothly produced. The comparable target products for 1f, 1g, and 1h were likewise converted in modest yields under these circumstances (3f–3h). The structure of 3g was verified using X-ray single crystal diffraction (CCDC 2304478). The system exhibited good compatibility with the chained tertiary alkyl NHPI esters as well, resulting in 48–57% yields of the desired products (3i–3l). Additionally, it was demonstrated that the similar compounds (3m–3o) could be obtained in reasonable yields from heterocyclic substituted tertiary alkyl NHPI esters. With sodium benzenesulfonothioates (3p–3t), tertiary alkyl NHPI esters produced from cyclohexyl could also be thiosulfonated. Remarkably, bioactive molecule-derived substrate like Gemfibrozil was also compatible to our conditions, the corresponding thiosulfonylation product 3u was obtained in 75% isolated yield. It is worth mentioning that substrates bearing different arene sulfonothioates were also competent coupling partners, and the desired product (3v–3ad) were obtained in moderate yields.

Reaction conditions: 1a (0.2 mmol), 2a (2 equiv, 0.4 mmol), 4CzIPN (2 mol%, 0.004 mmol), Cu(OAc)2 (20 mol%, 0.04 mmol), KHCO3 (2 equiv, 0.4 mmol), EA (2 mL) at room temperature (rt), 5 W blue LEDs, 12 h in N2. EA ethyl acetate.
Furthermore, the substrate scope for SS-thiosulfonates was subsequently investigated. As shown in Fig. 3, further study on the scope of the reaction revealed that various tertiary or secondary alkyl carboxylic acid derived esters could readily afford the SS-thiosulfonates. The product 4a derived from pivalic acid was obtained in 83% yield. When the substituents were tertiary cyclopropyl, cyclopentyl, cyclohexyl, pyran 1,3- dioxane or adamantyl, the yields of the desired products were moderate to good (4b–4g). It is indicated that various substituted tertiary NHPI esters and sodium benzenesulfonothioates were also well-suited for this transformation (4h–4l). Moreover, by increasing aliphatic chain length in tertiary carboxylic acids, the expected products were obtained in high yields (4m–4o). Interestingly, this catalytic system was also amenable to various NHPI esters derived from tertiary cyclohexanecarboxylic acids, and the corresponding sulfonothioates (4p–4u) were obtained in moderate to good yields (75-85%). Bicyclo[2,2,2]octyl group was introduced efficiently (4v) in 70% yield. Derived from pharmaceutical drugs such as Gemfibrozil, Ibuprofen and Flurbiprofen resulted in the formation of the corresponding disulfides in 59%–68% yields (4w–4y). Meantime, with the use of different sodium benzenesulfonothioates, the corresponding SS-thiosulfonates were constructed smoothly with yields of 63–91% (4z–4ah).

Reaction conditions: 1 (0.2 mmol), 2 (2 equiv, 0.4 mmol), 4CzIPN (2 mol%, 0.004 mmol), Cu(OTf)2 (20 mol%, 0.04 mmol), (BuO)2P(O)OH (2 equiv, 0.4 mmol), EA (2 mL) at room temperature (rt), 5 W blue LEDs, 12 h in N2. EA ethyl acetate.
To compare with tertiary alkyl NHPI esters, subsequently, the substrate range of secondary and primary alkyl NHPI esters for the synthesis of S-alkyl benzenesulfonothioates was explored and the results were summarized in Fig. 4. To our delight, with the secondary alkyl NHPI esters bearing various functionalized 3-, 4-, 5-, and 6-membered rings (including cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl), we were able to get the matching products (5a–5h) in moderate to good yields. Meanwhile, with sec-benzyl-substituted NHPI ester, the target product 5i was also obtained in 91% yield. We then switched to other sodium benzenesulfonothioates, and each of them was able to successfully render the appropriate target products (5j–5m) upon treatment in the standard conditions. The variety of substrates for primary alkyl NHPIs was then assessed. The corresponding benzenesulfonothioates (5n–5z) were produced in moderate to good yields via our thiosulfonation processes involving carboxylic acids such as adamantanes, thiophenes, ester groups, benzyl, alkyl, and so on. Of note, NHPI esters derived from various drug molecules and natural products, including Flurbiprofen, Lauric acid, Oleic acid, Mycophenolic acid, etc., were also good candidates for this transformation and the corresponding products (5aa–5ag) were successfully delivered under the standard conditions with 67–74% isolation yields.

Reaction conditions: a1 (0.2 mmol), 2 (2 equiv, 0.4 mmol), 4CzIPN (2 mol%, 0.004 mmol), Cu(OTf)2 (20 mol%, 0.04 mmol), (BuO)2P(O)OH (2 equiv, 0.4 mmol), EA (2 mL) at room temperature (rt), 5 W blue LEDs, 12 h in N2. EA ethyl acetate.
Downstream applications and transformations
To further demonstrate the synthetic value of this strategy, we carried out gram-scale synthesis of S-alkyl thiosulfonates (3a and 5d) as well as gram-scale preparation of SS-alkyl thiosulfonates (4z) under the corresponding standard conditions, and the efficiencies did not erode significantly (Fig. 5a). Furthermore, we also realized the one-pot strategy by directly using carboxylic acids as raw materials to achieve S-thiosulfonates and SS-thiosulfonates without presynthesis of NHPI esters in advance (Fig. 5b), which further demonstrates the universality and mild reaction conditions of our reaction. For S-thiosulfonates, pivalic acid, 3- or 6-membered ring acids were readily converted into the corresponding products (3a, 3b, 3d). Bicyclo[2.2.2]octane carboxylic acid was also transformed into the corresponding product in 35% yield (3e). 2-Methyl-2-phenylpropanoic acid (1f) was suitable candidate as well for this transformation. Similarly, when the same carboxylic acid was used, the SS-thiosulfonation could also be achieved by in-situ activation of carboxylic acids (4b, 4c, 4e, 4j, 4v). Furthermore, Gemfibrozil could be easily converted into the corresponding products (4w).

a Gram-scale synthesis. b One-pot synthesis of S-thiosulfonates and SS-thiosulfonates. Acid (0.22 mmol), NHPI (0.2 mmol), DCC (0.22 mmol), DMAP (0.02 mmol), rt, 5 h. aStandard condition A: 2 (2 equiv, 0.4 mmol), 4CzIPN (2 mol%, 0.004 mmol), Cu(OAc)2 (20 mol%, 0.04 mmol), KHCO3 (2 equiv, 0.4 mmol), EA (2 mL) at room temperature (rt), 5 W blue LEDs, 12 h in N2. bStardard condition B: 2 (2 equiv, 0.4 mmol), 4CzIPN (2 mol%, 0.004 mmol), Cu(OTf)2 (20 mol%, 0.04 mmol), (BuO)2P(O)OH (2 equiv, 0.4 mmol), EA (2 mL) at room temperature (rt), 5 W blue LEDs, 12 h in N2. EA ethyl acetate.
Subsequently, the synthetic transformations of benzenesulfonothioates and sulfur- sulfur sulfones were carried out (Fig. 6). S-Cyclohexyl benzenesulfonothioate 5d could be easily converted into secondary S-alkyl thiocarbamate 6 in 65% yield67. Using a developed procedure by Wang et al., a new organophotoredox thiolation reaction give thioether 736. According to the literature, S-cyclohexyl 4-methoxybenzothioate 8 could be readily acquired from 5d by visible light catalysis68.

A Transformation of S-alkyl thiosulfonates. B Transformation of SS-alkyl thiosulfonates. C Synthesis of steric hindered unsymmetric alkyl disulfides. D Application in the synthesis of 18. Reaction conditions: (a) NaI, tBuNC, iPrOH, air, 30 °C; (b) cyclohexyl NHPI ester, 4CzIPN, DIPEA, CH3CN, Blue LEDs, N2, rt; (c) aldehyde, 9,10-phenanthraquinone, Na2CO3, CH3CN, Blue LEDs, N2, rt; (d) benzyl azide, CuI, LitOBu, THF, N2, 40 °C; (e) 4-bromoaniline, tert-butyl nitrite, sodium ascorbate, MeCN, N2, 60 °C; (f) aryl boronic acid, CuSO4, NaHCO3, MeOH, N2, 60 °C; (g) 1-ethynyl-4-methoxybenzene, CuI, LitOBu, DCE, N2, 70 °C.
Interestingly, triazole 9 was prepared by copper(I)-catalyzed interrupted click reaction31. Using sodium ascorbate, unsymmetric disulfides 10 and 13 can be accessed from readily available anilines69. 11 was synthesized in good yield by using aryl boronic acid and SS-thiosulfonate70. A copper-catalyzed thiolation of terminal alkynes led to access alkynyl disulfides 1271. Then, using tertiary NHPI-derived RAEs as radical precursors to react with SS-thiosulfonates, more steric hindered unsymmetric alkyl disulfides (Fig. 6C, 14a–14i) were constructed successfully, of note, they are difficult to access in other known strategies36. As showcase the application of our benzenesulfonothioate strategy (Fig. 6D), we carried out the reaction between 15 and aminobutyric to generate aliphatic carboxylic acid 16, which was further activated in situ to afford NHPI ester, without further purification, the ester was subjected to the standard conditions to lead to thiosulfonate 17 via a one-pot method in 60% overall yield. Compound 17 was then employed to subsequent transformations to access Ajoene derivative 1872, which is a key intermediate for the synthesis of Ajoene.
Mechanism investigations
To gain insight into the mechanism of the reaction, radical scavengers (TEMPO, BHT or 1,1-diphenylalkene) were added under the standard conditions (Fig. 7a). The formation of compounds 3a and 4a were completely suppressed, which indicated that a single-electron-transfer radical process might be involved. The NHPI ester of cyclopropylacetic acid 22 reacted with compound 2a to form the ring-opening sulfonylation product 23 exclusively (Fig. 7b). These experiments demonstrated the intermediacy of alkyl radicals. To test the possibility of reaction intermediate, we subjected 3a to the standard conditions (Table 1, entry 10), as we speculated, 4b was obtained in 58% (Fig. 7c). When only 3 and 2 was added in the system without PC, Cu, and additive and light, the yield of 4b decreased.

a Radical capture experiment. b Radical-clock experiment. c The possibility of reaction intermediate.
Based on the aforementioned experimental investigation and related prior work73,74,75,76, we propose a plausible mechanism, as illustrated in Fig. 8. Initially, 4CzIPN forms the excited state 4CzIPN* under illumination. The 4CzIPN* undergoes a single electron transfer (SET) process with NHPI ester 1, resulting in the formation of the radical cation of 4CzIPN and the radical anion of NHPI ester A. The radical anion A is unstable and readily undergoes N-O bond cleavage, generating a carboxyl radical and a phthalate anion. Meanwhile, the carboxyl radical releases CO2, resulting in the formation of alkyl radical B. The radical cation of 4CzIPN then participates in SET with Cu(I) to complete its catalytic cycle, concurrently generating Cu(II). The Cu(II) species reacts with sulfide anion 2 as an oxidizing agent to form the intermediate C, which is subsequently captured by radical B to yield thiosulfinate ester 3. The benzenesulfonothioate 3 then undergoes nucleophilic addition with anion 2 to generate product 4 and D. The anion D is protonated to form sulfinic acid E.

Possible reaction mechanism of photocatalytic formation of S-alkyl thiosulfonates and SS-alkyl thiosulfonates.
In summary, we reported a visible light induced decarboxylative thiosulfonylation reaction. This method is straightforward, without requiring additional oxidants or reductants, and utilizes readily available and inexpensive starting materials, thus providing a robust pathway for the synthesis of unsymmetric S-thiosulfonates and SS-thiosulfonates, particularly with significant steric hindrance. This approach holds considerable promise for broad applications in thiosulfonylation. Through subsequent transformations, we synthesized mono-thiol, di-thiol, and unsymmetric aliphatic disulfides. Given the significance of benzenesulfonothioate, we are currently conducting a mechanism study and exploring more radical reactions involving benzenesulfonothioates in our laboratory.
Methods
General procedure for synthesis of tertiary S-benzyl benzenesulfonothioates
A mixture of 1 (0.2 mmol), 2 (0.4 mmol), 4CzIPN (0.004 mmol) and Cu(OAc)2 (0.04 mmol) were charged into a Schleck tube, then the air was removed, N2 was filled of Schleck tube and KHCO3 (0.4 mmol), EA (2 mL) is added the mixture. The mixture was stirred under irradiation from 5 W Blue LEDs. After the solvent was removed under reduced pressure, the residue was purified by silica gel chromatography using PE/EA (100:1) to afford the corresponding product.
General procedure for synthesis of sulfur–sulfur sulfone
A mixture of 1 (0.2 mmol), 2 (0.4 mmol), 4CzIPN (0.004 mmol) and Cu(OTf)2 (0.04 mmol) were charged into a Schleck tube, then the air was removed, N2 was filled of Schleck tube and (BuO)2P(O)OH (0.4 mmol), EA (2 mL) is added the mixture. The mixture was stirred under irradiation from 5 W Blue LEDs. After the solvent was removed under reduced pressure, the residue was purified by silica gel chromatography using PE/EA (100:1) to afford the corresponding product.
General procedure for synthesis of primary and secondary S-benzyl benzenesulfonothioates
A mixture of 1 (0.2 mmol), 2 (0.4 mmol), 4CzIPN (0.004 mmol) and Cu(OTf)2 (0.04 mmol) were charged into a Schleck tube, then the air was removed, N2 was filled of Schleck tube and (BuO)2P(O)OH (0.4 mmol), EA (2 mL) is added the mixture. The mixture was stirred under irradiation from 5 W Blue LEDs. After the solvent was removed under reduced pressure, the residue was purified by silica gel chromatography using PE/EA (100:1) to afford the corresponding product.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. All other data are available from the corresponding author upon request. The X-ray crystallographic coordinates for structures of 3g, 3ad, 4z and 5m reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2304478 (3g), 2322019 (3ad), 2160973 (4z) and 2180018 (5m). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
References
Deslauriers, R. & Somorjai, R. L. Internal rotations of side chains and backbone in luteinizing hormone-releasing hormone (LH-RH). Analysis of carbon-13 spin-lattice relaxation times. J. Am. Chem. Soc. 98, 1931–1939 (1976).
Lin, G.-Q., Xu, M.-H., Zhong, Y.-W. & Sun, X.-W. An advance on exploring N-tert-butanesulfinyl imines in asymmetric synthesis of chiral amines. Acc. Chem. Res. 41, 831–840 (2008).
D’Angelo, N. D. et al. Discovery and optimization of a series of benzothiazole phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) dual inhibitors. J. Med. Chem. 54, 1789–1811 (2011).
Jiang, C.-S., Müller, W. E. G., Schröder, H. C. & Guo, Y.-W. Disulfide- and multisulfide-containing metabolites from marine organisms. Chem. Rev. 112, 2179–2207 (2012).
Omann, L., Königs, C. D. F., Klare, H. F. T. & Oestreich, M. Cooperative catalysis at metal–sulfur bonds. Acc. Chem. Res. 50, 1258–1269 (2017).
Wang, N., Saidhareddy, P. & Jiang, X. Construction of sulfur-containing moieties in the total synthesis of natural products. Nat. Prod. Rep. 37, 246–275 (2020).
Kim, J. K., Bonicamp, J. & Caserio, M. C. Methoxymethyl cations. 2. Reactions with allylic ethers and sulfides in the gas phase. J. Org. Chem. 46, 4236–4242 (1981).
Mahieu, J.-P., Gosselet, M., Sebille, B. & Beuzard, Y. Synthesis of new thiosulfonates and disulfides from sulfonyl chlorides and thiols. Synth. Commun. 16, 1709–1722 (1986).
Chauhan, P., Mahajan, S. & Enders, D. Organocatalytic carbon-sulfur bond-forming reactions. Chem. Rev. 114, 8807–8864 (2014).
Musiejuk, M. & Witt, D. Recent developments in the synthesis of unsymmetrical disulfanes (disulfides). A review. Org. Prep. Proced. Int. 47, 95–131 (2015).
Wu, Q., Zhao, D., Qin, X., Lan, J. & You, J. Synthesis of di(hetero)aryl sulfides by directly using arylsulfonyl chlorides as a sulfur source. Chem. Commun. 47, 9188–9190 (2011).
Ge, W. & Wei, Y. Copper(I) iodide catalyzed 3-sulfenylation of indoles with unsymmetric benzothiazolyl-containing disulfides at room temperature. Synthesis 44, 934–940 (2012).
Yang, F.-L. & Tian, S.-K. Iodine-catalyzed regioselective sulfenylation of indoles with sulfonyl hydrazides. Angew. Chem. Int. Ed. 52, 4929–4932 (2013).
Rao, H. et al. K2S2O8/arenesulfinate: an unprecedented thiolating system enabling selective sulfenylation of indoles under metal-free conditions. RSC Adv. 4, 49165–49169 (2014).
Kumaraswamy, G., Raju, R. & Narayanarao, V. Metal- and base-free syntheses of aryl/alkylthioindoles by the iodine-induced reductive coupling of aryl/alkyl sulfonyl chlorides with indoles. RSC Adv. 5, 22718–22723 (2015).
Liu, C.-R. & Ding, L.-H. Byproduct promoted regioselective sulfenylation of indoles with sulfinic acids. Org. Biomol. Chem. 13, 2251–2254 (2015).
Weidner, J. P. & Block, S. S. Alkyl and aryl thiolsulfonates. J. Med. Chem. 7, 671–673 (1964).
Zefirov, N. S., Zyk, N. V., Beloglazkina, E. K. & Kutateladze, A. G. Thiosulfonates: synthesis, reactions and practical applications. Sulfur Rep. 14, 223–240 (1993).
Steudel, R. The chemistry of organic polysulfanes R-Sn-R (n > 2). Chem. Rev. 102, 3905–3946 (2002).
Kim, S., Kim, S., Otsuka, N. & Ryu, I. Tin-free radical carbonylation: thiol ester synthesis using alkyl allyl sulfone precursors, phenyl benzenethiosulfonate, and CO. Angew. Chem. Int. Ed. 44, 6183–6186 (2005).
Mampuys, P. et al. Sustainable three-component synthesis of isothioureas from isocyanides, thiosulfonates, and amines. Angew. Chem. Int. Ed. 53, 12849–12854 (2014).
Mai, S. & Song, Q. Divergent synthesis of disulfanes and benzenesulfonothioates bearing 2-aminofurans from N-tosylhydrazone-bearing thiocarbamates. Angew. Chem. Int. Ed. 56, 7952–7957 (2017).
Javitch, J. A., Li, X., Kaback, J. & Karlin, A. A cysteine residue in the third membrane-spanning segment of the human D2 dopamine receptor is exposed in the binding-site crevice. Proc. Natl Acad. Sci. 91, 10355–10359 (1994).
Gallardo-Godoy, A., Torres-Altoro, M. I., White, K. J., Barker, E. L. & Nichols, D. E. 1-Methylpyridinium-4-(4-phenylmethanethiosulfonate) iodide, MTS-MPP+, a novel scanning cysteine accessibility method (SCAM) reagent for monoamine transporter studies. Bioorg. Med. Chem. 15, 305–311 (2007).
Sugata, K. et al. Nucleotide-induced flexibility change in neck linkers of dimeric kinesin as detected by distance measurements using spin-labeling EPR. J. Mol. Biol. 386, 626–636 (2009).
Ge, C. et al. A thiol-thiosulfonate reaction providing a novel strategy for turn-on thiol sensing. Chem. Commun. 51, 14913–14916 (2015).
Kutateladze, A. G., Beloglazkina, E. K., Zyk, N. V. & Zefirov, N. S. S-tosylsulfene chloride—the first representative of a new class of sulfene halides. Russ. Chem. Bull. 41, 960–961 (1992).
Sotirova, A. et al. The importance of rhamnolipid-biosurfactant-induced changes in bacterial membrane lipids of bacillus subtilis for the antimicrobial activity of thiosulfonates. Curr. Microbiol. 65, 534–541 (2012).
Selvam, B., Mittal, S. & Shukla, D. Free energy landscape of the complete transport cycle in a key bacterial transporter. ACS Cent. Sci. 4, 1146–1154 (2018).
Ward, D. J., Van de Langemheen, H., Koehne, E., Kreidenweiss, A. & Liskamp, R. M. J. Highly tunable thiosulfonates as a novel class of cysteine protease inhibitors with anti-parasitic activity against Schistosoma mansoni. Bioorg. Med. Chem. 27, 2857–2870 (2019).
Wang, W., Peng, X., Wei, F., Tung, C.-H. & Xu, Z. Copper(I)-catalyzed interrupted click reaction: synthesis of diverse 5-hetero-functionalized triazoles. Angew. Chem. Int. Ed. 55, 649–653 (2016).
Ghiazza, C. et al. Visible-light-mediated metal-free synthesis of trifluoromethylselenolated arenes. Angew. Chem. Int. Ed. 57, 11781–11785 (2018).
Li, J., Zhu, D., Lv, L. & Li, C.-J. Radical difluoromethylthiolation of aromatics enabled by visible light. Chem. Sci. 9, 5781–5786 (2018).
Qi, J., Wei, F., Huang, S., Tung, C.-H. & Xu, Z. Copper(I)-catalyzed asymmetric interrupted kinugasa reaction: synthesis of α-thiofunctional chiral β-lactams. Angew. Chem. Int. Ed. 60, 4561–4565 (2021).
Xu, B., Wang, D., Hu, Y. & Shen, Q. Silver-catalyzed ring-opening difluoromethylthiolation/trifluoromethylthiolation of cycloalkanols with PhSO2SCF2H or PhSO2SCF3. Org. Chem. Front. 5, 1462–1465 (2018).
Dong, Y. et al. Organophotoredox-catalyzed formation of alkyl-aryl and -alkyl C-S/Se bonds from coupling of redox-active esters with thio/selenosulfonates. Org. Lett. 22, 9562–9567 (2020).
Li, J. et al. Visible-light-promoted cross-coupling reactions of 4-Alkyl-1,4-dihydropyridines with thiosulfonate or selenium sulfonate: a unified approach to sulfides, selenides, and sulfoxides. Org. Lett. 22, 4908–4913 (2020).
Wu, Z. & Pratt, D. A. A divergent strategy for site-selective radical disulfuration of carboxylic acids with trisulfide-1,1-dioxides. Angew. Chem. Int. Ed. 60, 15598–15605 (2021).
Zhou, X., Pyle, D., Zhang, Z. & Dong, G. Deacylative thiolation by redox-neutral aromatization-driven C-C fragmentation of ketones. Angew. Chem. Int. Ed. 62, e202213691 (2023).
Wu, H. et al. Construction of C-S and C-Se bonds from unstrained ketone precursors under photoredox catalysis. Angew. Chem. Int. Ed. 63, e202314790 (2024).
Zhu, D., Shao, X., Hong, X., Lu, L. & Shen, Q. PhSO2SCF2H: a shelf-stable, easily scalable reagent for radical difluoromethylthiolation. Angew. Chem. Int. Ed 55, 15807–15811 (2016).
Li, H., Shan, C., Tung, C.-H. & Xu, Z. Dual gold and photoredox catalysis: visible light-mediated intermolecular atom transfer thiosulfonylation of alkenes. Chem. Sci. 8, 2610–2615 (2017).
Gadde, K. et al. Thiosulfonylation of unactivated alkenes with visible-light organic photocatalysis. ACS Catal. 10, 8765–8779 (2020).
Peng, Z., Yin, H., Zhang, H. & Jia, T. Regio- and stereoselective photoredox-catalyzed atom transfer radical addition of thiosulfonates to aryl alkynes. Org. Lett. 22, 5885–5889 (2020).
Lü, S., Wang, Z., Gao, X., Chen, K. & Zhu, S. 1,2-Difunctionalization of acetylene enabled by light. Angew. Chem. Int. Ed. 62, e202300268 (2023).
Ren, X. et al. Access to polysulfides through photocatalyzed dithiosulfonylation. Angew. Chem. Int. Ed. 62, e202302199 (2023).
Chen, Y. et al. Nickel(ii)/TPMPP catalyzed reductive coupling of oxalates and tetrasulfides: synthesis of unsymmetric disulfides. Org. Chem. Front. 9, 4962–4968 (2022).
Wang, F., Chen, Y., Rao, W., Ackermann, L. & Wang, S.-Y. Efficient preparation of unsymmetrical disulfides by nickel-catalyzed reductive coupling strategy. Nat. Commun. 13, 2588 (2022).
Cao, J.-M. et al. Simultaneous preparation of sulfides/selenides and sulfones via synergistic nickel-catalyzed reductive coupling and SN2 reaction. Org. Lett. 25, 9207–9212 (2023).
Fujiki, K., Tanifuji, N., Sasaki, Y. & Yokoyama, T. New and facile synthesis of thiosulfonates from sulfinate/disulfide/I2 system. Synthesis 2002, 0343–0348 (2002).
Liang, G. et al. NBS-promoted sulfenylation of sulfinates with disulfides leading to unsymmetrical or symmetrical thiosulfonates. Chin. J. Chem. 30, 1611–1616 (2012).
Pham, H. T., Nguyen, N.-L. T., Duus, F. & Luu, T. X. T. Ultrasound-accelerated synthesis of asymmetrical thiosulfonate S-esters by base-promoted reaction of sulfonyl chlorides with thiols. Phosphorus Sulfur Silicon Relat. Elem. 190, 1934–1941 (2015).
Gui, Y., Qiu, L., Li, Y., Li, H. & Dong, S. Internal activation of peptidyl prolyl thioesters in native chemical ligation. J. Am. Chem. Soc. 138, 4890–4899 (2016).
Gong, K., Zhou, Y. & Jiang, X. From symmetrical tetrasulfides to trisulfide dioxides via photocatalysis. Green Chem. 23, 9865–9869 (2021).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Cheung, K. P. S., Sarkar, S. & Gevorgyan, V. Visible light-induced transition metal catalysis. Chem. Rev. 122, 1543–1625 (2022).
Bellotti, P. & Glorius, F. Strain-release photocatalysis. J. Am. Chem. Soc. 145, 20716–20732 (2023).
Ham, R., Nielsen, C. J., Pullen, S. & Reek, J. N. H. Supramolecular coordination cages for artificial photosynthesis and synthetic photocatalysis. Chem. Rev. 123, 5225–5261 (2023).
Xu, G.-Q., Wang, W. D. & Xu, P.-F. Photocatalyzed enantioselective functionalization of C(sp3)-H bonds. J. Am. Chem. Soc. 146, 1209–1223 (2024).
He, J. et al. Catalytic decarboxylative radical sulfonylation. Chem 6, 1149–1159 (2020).
Mao, R., Balon, J. & Hu, X. Decarboxylative C(sp3)-O cross-coupling. Angew. Chem. Int. Ed. 57, 13624–13628 (2018).
Mao, R., Balon, J. & Hu, X. Cross-coupling of alkyl redox-active esters with benzophenone imines: tandem photoredox and copper catalysis. Angew. Chem. Int. Ed. 57, 9501–9504 (2018).
Wang, C. et al. Visible-light-driven, copper-catalyzed decarboxylative C(sp3)-H alkylation of glycine and peptides. Angew. Chem. Int. Ed. 57, 15841–15846 (2018).
Dong, X.-Y. et al. A general asymmetric copper-catalysed Sonogashira C(sp3)-C(sp) coupling. Nat. Chem. 11, 1158–1166 (2019).
Guo, Y., Luo, Y., Mu, S., Xu, J. & Song, Q. Photoinduced decarboxylative phosphorothiolation of N-hydroxyphthalimide esters. Org. Lett. 23, 6729–6734 (2021).
Tian, Y. et al. A general copper-catalysed enantioconvergent C(sp3)-S cross-coupling via biomimetic radical homolytic substitution. Nat. Chem. 16, 466–475 (2023).
Mampuys, P. et al. Iodide-catalyzed synthesis of secondary thiocarbamates from isocyanides and thiosulfonates. Org. Lett. 18, 2808–2811 (2016).
Zhang, Y. et al. Organocatalytic transformation of aldehydes to thioesters with visible light. Chem. Eur. J. 25, 8225–8228 (2019).
Chen, S. et al. Sandmeyer-type reductive disulfuration of anilines. Org. Lett. 23, 7428–7433 (2021).
Wang, W., Lin, Y., Ma, Y., Tung, C.-H. & Xu, Z. Cu-catalyzed electrophilic disulfur transfer: synthesis of unsymmetrical disulfides. Org. Lett. 20, 3829–3832 (2018).
Li, J., Li, M., Duan, X. & Song, W. Copper-catalyzed thiolation of terminal aromatic alkynes to access alkynyl disulfides. Tetrahedron Lett. 61, 152256 (2020).
Hunter, R., Kaschula, C., Stellenboom, N., Cotton, J. & Parker, M. I. New excursions into the synthesis and medicinal chemistry of the disulfide bond. Phosphorus Sulfur Silicon Relat. Elem. 188, 1497–1507 (2013).
Wang, D., Zhu, N., Chen, P., Lin, Z. & Liu, G. Enantioselective decarboxylative cyanation employing cooperative photoredox catalysis and copper catalysis. J. Am. Chem. Soc. 139, 15632–15635 (2017).
Zhao, W., Wurz, R. P., Peters, J. C. & Fu, G. C. Photoinduced, copper-catalyzed decarboxylative C-N coupling to generate protected amines: an alternative to the curtius rearrangement. J. Am. Chem. Soc. 139, 12153–12156 (2017).
Xia, H.-D. et al. Photoinduced copper-catalyzed asymmetric decarboxylative alkynylation with terminal alkynes. Angew. Chem. Int. Ed. 59, 16926–16932 (2020).
Yi, X., Mao, R., Lavrencic, L. & Hu, X. Photocatalytic decarboxylative coupling of aliphatic N-hydroxyphthalimide esters with polyfluoroaryl nucleophiles. Angew. Chem. Int. Ed. 60, 23557–23563 (2021).
Acknowledgements
Financial support from the National Key Research & Development Program of China (2023YFF0723900 to Q.S.), National Natural Science Foundation of China (22271105 to Q.S., 21931013 to Q.S.), Natural Science Foundation of Fujian Province (2022J02009 to Q.S.), Promotion Program for Young and Middle-aged Teacher in Science and Technology Research of Huaqiao University (ZQN-705 to J.X.) and Open Research Fund of School of Chemistry and Chemical Engineering, Open Research Fund of School of Chemistry and Chemical Engineering (Henan Normal University to Q.S.) are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Q.S. and J.X. conceived and directed the project. Y.G., G.L. and M.Z. performed experiments and Y.G. prepared the Supplementary Information. G.L. and M.Z. helped collecting some new compounds and analyzing the data. Q.S. and J.X. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chaozhong Li, Shun-Yi Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Y., Lin, G., Zhang, M. et al. Photo-induced decarboxylative C-S bond formation to access sterically hindered unsymmetric S-alkyl thiosulfonates and SS-alkyl thiosulfonates. Nat Commun 15, 7313 (2024). https://doi.org/10.1038/s41467-024-51334-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51334-5
