
Abstract
The CRISPR-based detection methods have been widely applied, yet they remain limited by the non-universal nature of one-pot diagnostic approaches. Here, we report a universal one-pot fluorescent method for the detection of epidemic pathogens, delivering results within 15-20 min. This method uses heparin sodium to precisely tunes the cis-cleavage capability of Cas12 via interference with the Cas12a-crRNA binding process, thereby generating significant fluorescence due to the accumulation of isothermal amplification products. Additionally, this universal assay accommodates both classic and suboptimal PAMs, as well as various Cas12a subtypes such as LbCas12a, AsCas12a, and AapCas12b. Such a robust method demonstrates sensitivity and specificity exceeding 95% in the detection of monkeypox pseudovirus, influenza A virus, and SARS-CoV-2 from saliva or wastewater samples, when compared with qPCR or RT-qPCR. Moreover, the cost of heparin sodium per thousand uses is $0.01 to $0.04 only. Collectively, this universal and fast one-pot approach based on heparin sodium offers potential possibilities for point-of-care testing.
Similar content being viewed by others

Introduction
In recent years, large-scale epidemics have occurred frequently caused by viruses such as Zika virus1, Ebola virus2, SARS-CoV-23, and monkeypox (mpox) virus4. Notably, SARS-CoV-2 led to approximately 16 million deaths in two years, reducing the global average life expectancy by 1.6 years5. In this context, rapid and efficient monitoring systems are crucial for mitigating the spread of pandemics6. However, traditional quantitative polymerase chain reaction (qPCR) based methods face limitations due to the lengthy response time (4–24 h), encompassing sampling, transportation, and detection processes, as well as the high costs associated with laboratory construction and operation7,8,9. Thus, developing rapid, sensitive, and simple point-of-care testing (POCT) methods is essential for promoting home and on-site diagnostics and controlling disease transmission.
POCT methods, represented by antigen tests and isothermal amplification techniques such as recombinase polymerase amplification (RPA), loop-mediated isothermal amplification (LAMP), and rolling circle amplification (RCA) have been widely employed in detecting epidemic pathogens10,11,12. These rapid diagnostic technologies can deliver results within 10-30 min13,14. However, their limited sensitivity and relatively high false-negative rates constrain their broader application6,15. Clustered regularly interspaced short palindromic repeats (CRISPR) technology, initially central to gene editing, has ventured into molecular diagnostics after the discovery of the trans-cleavage activity of Cas12, Cas13, Cas12a2, and Cas916,17,18,19,20. Using Cas12 as an example, it specifically recognizes and cleaves target nucleic acids, followed by indiscriminate cleavage of surrounding single-stranded DNA (ssDNA)21. CRISPR-diagnostics (Dx) methods leverage this mechanism by amplifying nucleic acids through RPA, LAMP, or RCA, followed by the introduction of Cas12, crRNA, and ssDNA probes with fluorescent/quenching groups for detection22,23,24. This integration of CRISPR systems with isothermal amplification has markedly improved the sensitivity and specificity of pathogens detection25.
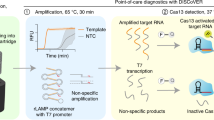
Early CRISPR-Dx such as DNA endonuclease-targeted CRISPR trans reporter (DETECTR) and one-HOur Low-cost Multipurpose highly Efficient System (HOLMES) required a two-step process involving initial amplification followed by the addition of CRISPR components17,26. However, the implementation of two steps increases operational complexity, extends response time, and lead to an elevated risk of nucleic acid aerosol contamination27. To address these challenges, one-pot detection systems have emerged, enabling simultaneous high-efficiency amplification and CRISPR-mediated detection within a single tube22. A key objective in developing universal one-pot detection systems is to prevent the cleavage activity of Cas12 from interfering with isothermal amplification processes6,28. Some studies have utilized spatial isolation of reagents or employed glycerol and sucrose to facilitate phase separation of amplification and detection solutions, thereby achieving one-pot detection29,30,31. Recently, light-controlled crRNA strategies have been ingeniously applied to lock and unlock the cleavage activity of Cas1227,28,32. Despite significant efforts, the aforementioned one-pot detection systems that either prevent Cas12 contacting with the amplification system or directly switch the cleavage activity of Cas12 still result in longer detection times and operational inconvenience. It is reported that targeting suboptimal PAM sequences with LbCas12a-crRNA can attenuate its cis-cleavage activity, enabling one-pot detection23. This is a promising approach due to its simplicity and rapid detection. However, this strategy requires designing crRNAs targeting multiple suboptimal PAMs for limit of detection (LoD) testing and cannot be generalized to canonical PAM sequences and AapCas12b6,33. The aforementioned studies demonstrate that regulating Cas12 cleavage activity is crucial for developing universally applicable one-pot detection methods.
In this work, we discover that the cleavage activity of Cas12a could be regulated by varying concentrations of heparin sodium via interference with the Cas12a-crRNA binding process. With this finding, we develop a universal one-pot detection method named SURVEY (heparin Sodium Used for Rapid Viral dEtection and analYsis). By simply adjusting the concentration of heparin sodium, SURVEY can accommodate canonical or suboptimal PAM sequences, with a LoD as low as 0.6 cp/μL. This method is compatible with Cas12 subtypes such as AsCas12a and AapCas12b, demonstrating high versatility. Additionally, the detection process takes only 15-20 min, making it 2-3 times faster than traditional CRISPR-Dx methods. In tests targeting mpox pseudovirus, influenza A virus, and SARS-CoV-2, SURVEY exhibited over 95% sensitivity and specificity in saliva and wastewater samples, indicating its broad applicability for various pathogen detections. Therefore, our method addresses a longstanding challenge in CRISPR-Dx by adding heparin sodium, providing a universal one-pot detection approach for point-of-care testing (POCT).
Results
Regulation of LbCas12a cleavage activity by adjusting the heparin sodium concentration
In CRISPR diagnostics, LbCas12a is widely recognized for its exceptional cis- and trans-cleavage activities17. Interestingly, we discovered that heparin sodium greatly inhibited LbCas12a’s cleavage activities, as initially revealed by plasmid and DNA fragment cleavage experiments. (Fig. 1a-b and Supplementary Fig. 1). The crRNAs were designed targeting the f3l and b6r genes of the mpox, which recognize the canonical PAM sequence (TTTA)34. These genes were pre-cloned into the pYYDT plasmid. Subsequently, these plasmids were mixed with LbCas12a and crRNAs to assess the cis-cleavage activity of the Cas12a-crRNA complex in the presence of 2 μg/mL heparin sodium. In the absence of heparin sodium, the plasmids were cleaved from supercoiled to linear states, consistent with the cleavage results produced by the endonuclease BsaI. However, upon the addition of 2 μg/mL heparin sodium, the state of plasmids remained in a state similar to that of the negative control (Fig. 1b). Furthermore, when using amplified and purified gene fragments of f3l and b6r as targets, dosing heparin sodium preserved the integrity of the targets (Supplementary Fig. 1). These findings suggest that heparin sodium largely inhibited the cis-cleavage activity of Cas12a.

a Schematic diagram of heparin sodium regulating Cas12a cleavage activity. b Cis-cleavage activity characterized using a plasmid containing the f3l and b6r genes. This plasmid also includes a BsaI restriction site. c Fluorescence detection of LbCas12a cis-cleavage activity in the presence of 2 µg/mL heparin sodium, with crRNA targeting f3l or b6r. d, e Fluorescent characterization of cis-cleavage at various concentrations of heparin sodium. The aggregated f3l and b6r fluorescent probe templates are cleaved to generate fluorescence. f Fluorescence detection of LbCas12a trans-cleavage activity in the presence of 2 µg/mL heparin sodium, with crRNA targeting f3l or b6r. g, h Fluorescent characterization of trans-cleavage at various concentrations of heparin sodium, with FAM-CCCCCCCC-BHQ1 being cleaved to generate fluorescence. Data are shown as mean ± s.d. for n = 3 biologically independent samples.
To further characterize the inhibitory effect of heparin sodium on the cis-cleavage activity of Cas12a, we labeled the 3’ end of the 35 nt non-target strand (NTS) with a quenching group (BHQ1) and the 5’ end of target strand (TS) with a fluorescent group (FAM). The TS and NTS strands, through polymerization forming T-DNA, were utilized to activate the cis-cleavage activity of the Cas12a. When the T-DNA was cleaved into two segments at 37 °C, the DNA strands unwound at the same temperature, causing separation of the fluorescent and quenching groups, thereby generated a significant fluorescence signal (Supplementary Fig. 2a). In the presence of heparin sodium, a notable suppression of the fluorescence signal was observed, further confirming the significant inhibitory effect of heparin sodium on the cis-cleavage activity of Cas12a (Fig. 1c and Supplementary Fig. 3a). Subsequently, we tried to regulate the cis-cleavage activity of Cas12a by adjusting the heparin sodium concentration. The results indicate that, as the heparin sodium concentration increased from 0.2 μg/mL to 0.5 μg/mL, the cis-cleavage activity of Cas12a was progressively inhibited (Fig. 1d, e).
These findings prompted a further investigation into the extent of inhibition by heparin sodium on the trans-cleavage activity of Cas12a (Supplementary Fig. 2b). After adding a fluorescent probe (FAM-CCCCCCCC-BHQ1) to the Cas12a-crRNA system containing 10 nM target DNA, we observed rapid and significant fluorescence signals without the addition of heparin sodium. However, upon dosing 2 μg/mL heparin sodium, the fluorescence signals were similar to that of the control, indicating that heparin sodium greatly inhibited Cas12a’s trans-cleavage activity (Fig. 1f). Furthermore, heparin sodium at 0.2 μg/mL was the minimum inhibitory concentration, with nearly complete inhibition of trans-cleavage activities at 0.5 μg/mL (Fig. 1g, h). When adjusting the concentration of substrate DNA, the required concentration of heparin sodium to inhibit trans-cleavage activity also varied accordingly (Supplementary Fig. 4a-f). At a substrate DNA concentration of 5 nM, only 0.2 μg/mL of heparin sodium was needed to effectively inhibit trans-cleavage activity. Meanwhile, when the substrate DNA concentration was increased to 50 nM, a heparin sodium concentration of 2 μg/mL was required to achieve complete inhibition. However, the precise mechanism by which heparin sodium inhibited the cleavage activity of Cas12a remained unclear. It is uncertain whether the inhibition disrupted the binding of Cas12a to crRNA, the interaction of the Cas12a-crRNA complex with T-DNA, or interfered with the activated Cas12a’s ability to cleave ssDNA.
Inhibition mechanism of Cas12a cleavage activity by heparin sodium
To investigate the effects of heparin sodium on the cis- and trans- cleavage activities of Cas12a, we conducted a comprehensive analysis across various stages of cleavage. This analysis included examining the interaction between Cas12a and crRNA, the cleavage of target DNA by the Cas12a-crRNA complex, or the cleavage reactions within the Cas12a-crRNA-T-DNA ternary complex using a fluorescent probe in the presence or absence of heparin sodium (Fig. 2a). First, when Cas12a bound with crRNA, the immediate addition of the 0.5 μg/mL heparin sodium before introducing T-DNA and the probe largely reduced the fluorescence signal (Fig. 2b, c). Under the latter two conditions, all binary and ternary complexes were first incubated for 20 min to ensure full binding. Additional components were then dosed to observe the effect of heparin sodium on the resulting fluorescence signals. However, dosing 0.5 μg/mL heparin sodium did not affect the generated fluorescence signals under the latter two conditions (Fig. 2b, c). These results suggest that heparin sodium primarily acted by interfering with the binding process between Cas12a and crRNA. Furthermore, the results also indicate that heparin sodium affected the cis-cleavage activity of the Cas12a-crRNA complex, which in turn indirectly impacted its trans-cleavage activity.

a Schematic representation of the three potential stages of Cas12a activity inhibited by heparin sodium. b, c Impact of heparin sodium on Cas12a cleavage activity at three distinct stages when crRNA targets f3l (b) or b6r (c). d The binding affinity of crRNA for Cas12a in the presence of heparin sodium using EMSA. e ΔG of crRNA, heparin sodium, and S-free heparin with LbCas12a. f Root Mean Square Deviation (RMSD) of LbCas12a in different simulation systems. g–k, Electrostatic potential analysis between Cas12a and crRNA (g); Cas12a and heparin (h); Cas12a, crRNA, and heparin (i); Cas12a and S-free heparin (j); Cas12a, crRNA, and S-free heparin (k). Data are shown as mean ± s.d. for n = 3 biologically independent samples.
Heparin sodium is a highly sulfated linear glycosaminoglycan with a strong negative charge effect. Therefore, we hypothesized that heparin sodium might interfere with the binding of Cas12a to crRNA, possibly due to its high content of negatively charged groups (HSO3- and SO42-), thereby competing with crRNA for the binding sites on the Cas12a (Supplementary Fig. 5). To test this hypothesis, the binding affinity of Cas12a to crRNA was verified through the electrophoretic mobility shift assay (EMSA). If the crRNA bound strongly to Cas12a, the Cas12a-crRNA band would appear above the PAGE lane, and vice versa, the crRNA band would appear below the lane. The results show that heparin sodium inhibited the binding of Cas12a with crRNA, resulting in a distinct band below the PAGE lane (Fig. 2d).
Subsequently, we utilized molecular dynamics (MD) simulation to elucidate the binding processes of crRNA and heparin with Cas12a individually, as well as the interaction among heparin, crRNA, and Cas12a when all three were present. In all the binary and ternary systems, the distances between Cas12a and crRNA or heparin decreased rapidly over the 100-ns simulation (Supplementary Fig. 6a, b), suggesting there was strong attractive interaction between Cas12a and crRNA or heparin. Such a conclusion was supported by their negative Gibbs free energy (ΔG) (Fig. 2e). Specifically, the Cas12-heparin exhibited the lowest ΔG. When the anionic groups (sulfonic/sulfate) on heparin were removed to form S-free-heparin, the ΔG of Cas12a-S-free-heparin was comparable to that of Cas12a-crRNA (Fig. 2e and Supplementary Fig. 7). These results suggest that the anion groups of heparin contributed largely to the interfacial attractive interaction. According to the trajectory, all the simulation systems approached stable at the last 20 ns (Fig. 2f). Hence, the complex structures were extracted for electrostatic potential analysis. The positive and negative areas of LbCas12a were unevenly distributed and the other small molecules were all negatively charged (Supplementary Fig. 8). In Cas12a-crRNA system, negatively charged crRNA partially occupied its binding site with positive surroundings and a complete incorporation required longer simulation time (Fig. 2g and Supplementary Fig. 8)35. Driven by electrostatic attraction, heparin traversed the active site of Cas12a and occupied the groove that bound crRNA in Cas12a-crRNA systems (Fig. 2h). When crRNA and heparin coexisted in the simulation box, heparin outperformed crRNA in occupying the groove owing to its lower ΔG (Fig. 2i). Such a phenomenon was not observed when crRNA competed with S-free heparin (Fig. 2j, k and Supplementary Fig. 9). Therefore, these MD simulation results indicate that heparin sodium, with its negatively charged groups, interfered the correct binding of crRNA with Cas12a, thus affecting the formation of the correct crRNA-Cas12a binary complex.
Inspired by the results of the electrostatic potential analysis, we investigated whether another glycosaminoglycan molecule with negatively charged groups, chondroitin sulfate, also had a similar inhibitory effect (Supplementary Fig. 10a, b). The results show that chondroitin sulfate could also significantly inhibit Cas12a’s cleavage activity, albeit at concentrations much higher than those for heparin sodium (Supplementary Fig. 10c, d). These findings suggest that glycosaminoglycan molecules with negative charge effects might be capable of inhibiting Cas12a’s cleavage activity.
One-pot detection design by leveraging heparin sodium inhibitory effect
By effectively regulating the cis-cleavage activity of Cas12a, we tried to explore a one-pot detection strategy (RPA-Cas12a) in the CRISPR-Dx system. In this experiment, the f3l and b6r genes of the mpox were selected as detection targets, and crRNAs were designed for these genes using the canonical PAM sequence (TTTA), integrated with RPA (Fig. 3a). In the absence of heparin sodium, the RPA-Cas12a combination did not produce significant fluorescence signals within the target gene concentration range of 0.1 aM to 10 fM (Fig. 3b, e and Supplementary Fig. 11a,c). Even when the reaction time was extended to 12 h, no significant fluorescence signal was observed (Supplementary Fig. 12a, b). To address this issue, we hypothesized that adding an appropriate amount of heparin sodium to the one-pot detection system might enhance the generation of fluorescence signals. Considering that the previous inhibition experiments were conducted with a simple buffer system, whereas the one-pot detection involved a more complex composition, the inhibition concentrations were examined using two additional complex buffers (previously employed buffer termed as Buffer-1). We found that Buffer-2 required more than 10 μg/mL of heparin sodium to completely inhibit its cleavage activity (Supplementary Fig. 13a). Buffer-3 (the New England Biolabs buffer-rCutSmart buffer) required 50-100 μg/mL of heparin sodium (Supplementary Fig. 13b). These results suggest that, if the used buffer had a greater Cas12a cleavage activity, a higher heparin sodium concentration would be needed to inhibit its activity. Therefore, we initially introduced 50 μg/mL of heparin sodium into the one-pot detection system. The RPA-Cas12a assay exhibited significant fluorescence signals across a target concentration range from 1 aM (≈0.6 copies/μL, volume=30 μL) to 10 fM (≈6000 copies/μL, volume=30 μL), enhancing detection sensitivity by more than 10,000-fold compared to the system without heparin sodium (Fig. 3c, f and Supplementary Fig. 11b, d). The LoD of the one-pot assay based on heparin sodium was comparable to that of quantitative PCR (qPCR) and the two-step method (Fig. 3d, g and Supplementary Fig. 14a, b). To assess the stability of the LoD, we conducted 20 replicate assays for the f3l and b6r genes at a concentration of 1 aM. The results demonstrate that the heparin sodium-mediated one-pot method reliably detected 1 aM of the f3l and b6r genes (Supplementary Fig. 15a, b). Furthermore, we also assessed the impact of the concentrations of heparin sodium, Cas12a, and crRNA on the detection efficiency in the RPA-Cas12a detection system. Results indicate that the optimal detection efficiency was achieved when the concentration of heparin sodium was within a range of 40-60 μg/mL, while the concentrations of Cas12a and crRNA did not largely affect the detection efficiency (Supplementary Fig. 16a–c). The results indicate significant differences in the heparin sodium concentrations required for inhibition across different buffers, highlighting the need to optimize its concentration when developing one-pot detection methods.

a, b Schematic of the one-pot detection method, where RPA amplification and Cas12a-mediated fluorescence detection occur simultaneously within a single tube. b–d Characterization of the LoD of the RPA-Cas12a one-pot detection method without (b) and with 40 µg/mL heparin sodium (c) and the qPCR (d), targeting the f3l gene at concentrations of 0, 0.1, 1, 10, 100, 1000, 10000 aM. e–g LoD characterization of the RPA-Cas12a one-pot method targeting b6r without (e) and with the 40 µg/mL heparin sodium (f) and the qPCR (g). h, i At a target concentration of 10 fM, with Cas12a-crRNA (100 nM) targeting f3l (h) or b6r (i), the content of RPA amplification products at various times were analyzed in agarose gels. Samples without heparin sodium or Cas12a during the RPA amplification process were used as the controls. Data are shown as mean ± s.d. for n = 3 biologically independent samples. The schematics shown in Fig. 3a were created by figdraw.com.
We further analyzed the amplification of target nucleic acids in the RPA-Cas12a system. To assess the amplification profiles under different conditions, the reactions were respectively terminated at 0, 2, 4, 6, 8, 12, 16, and 20 min. The resulting products were then analyzed by agarose gels in three distinct setups: RPA-heparin sodium without Cas12a, RPA-Cas12a without heparin sodium, and RPA-Cas12a with heparin sodium. Results show that in the absence of heparin sodium, nucleic acid amplification was hindered due to the high cis-cleavage activity of Cas12a (Fig. 3h, i). However, after dosing heparin sodium, nucleic acid amplification greatly increased due to the diminished cis-cleavage activity of Cas12a, although it did not reach the levels achieved with the RPA amplification system alone (Fig. 3h, i). Simultaneously, we evaluated the effect of heparin sodium concentration on the RPA reaction (Supplementary Fig. 17). The results indicate that when the heparin sodium concentration exceeded 80 µg/mL, the RPA reaction rate was inhibited, which was consistent with our above findings (Supplementary Fig. 16a and Supplementary Fig. 17). Subsequently, we tested the effect of 40 μg/mL heparin sodium on RPA at target concentrations of 1 fM, 100 aM, 10 aM, 1 aM, 0.1 aM, and 0 aM. The results show that under 40 μg/mL heparin sodium, RPA efficiency was only slightly affected compared to conditions without heparin sodium, with the LoD remaining at 1 aM (Supplementary Fig. 18). These findings reveal that during trace nucleic acid detection, the accumulation of amplification products occurs because Cas12a’s cleavage activity is inferior to the amplification capacity of RPA. We hypothesize that as amplification products accumulate, Cas12a’s cleavage activity gradually recovers. Therefore, we further tested the concentrations of heparin sodium required to inhibit Cas12a’s cleavage activity at target concentrations of 10, 50, and 100 nM (Supplementary Fig. 19a–f). The results indicate that as the target concentration increased, 40 μg/mL heparin sodium could no longer effectively suppress Cas12a’s cleavage activity, leading to significant fluorescent signals. In addition, a one-pot assay was also conducted using chondroitin sulfate as substitutes for heparin sodium. Chondroitin sulfate was found to produce noticeable fluorescence at concentrations above 100 μg/mL (Supplementary Fig. 20). This reinforces that the core of one-pot detection lied in the precise downregulation of the cleavage activity of Cas12a.
The classical PAMs for LbCas12a are TTTV (where V represents A, G, or C), with the enzyme exhibiting its highest cleavage activity when recognizing these classical PAMs36. The above results indicate that dosing heparin sodium enabled the detection of trace nucleic acids in a one-pot assay when the PAM was TTTA (Fig. 3d, e). Furthermore, the two remaining classical PAMs, TTTG and TTTC, were also characterized in the one-pot assay. We found that heparin sodium-mediated one-pot detection was achievable when targeting TTTG or TTTC (Fig. 4a, b and Supplementary Fig. 21a–d). Previous studies show that some suboptimal PAMs were suitable for one-pot detection23. Therefore, we tested three suboptimal PAMs (CTTT, ATTA, TTAA). The results indicate that heparin sodium enhanced detection efficiency in the one-pot assay when suboptimal PAMs were recognized (Fig. 4c–e). Subsequently, we conducted a comprehensive assessment about the effect of low concentrations of Cas12a on the one-pot detection method. The results indicate that reducing the Cas12a concentration did not produce significant fluorescence signals (Supplementary Fig. 22a). However, upon dosing heparin sodium to these systems, substantial fluorescence signals were observed (Supplementary Fig. 22b). To further examine the detection performance of low-concentration Cas12a, we conducted one-pot detection using commercial Cas12a (Tolobio, Co., China) and synthetic crRNA (Tolobio, Co., China), and the results were consistent with our findings (Supplementary Fig. 23a, b). Additionally, we further investigated the impact of crRNAs with different targeting lengths (spacer lengths = 10, 15, 17–20, 25 and 30 nt) on one-pot detection, and the results demonstrate that significant fluorescence signals were generated only in the presence of heparin sodium (Supplementary Fig. 24a–i). These findings suggest that heparin sodium-mediated Cas12a one-pot detection extended its applicability to both classic and suboptimal PAMs, varying Cas protein concentrations, and different crRNA targeting lengths, thereby significantly enhancing the versatility of one-pot detection methods.

a Validation of the SURVEY method when targeting f3l at 1 fM (1000 aM) with classical PAMs (TTTC and TTTG). bValidation of the SURVEY method when targeting b6r at 1 fM with classical PAMs (TTTC and TTTG). c–e Validation of the SURVEY method when targeting f3l or b6r with suboptimal PAMs (CTTT (c), ATTA (d) and TTAA (e)). f Schematic illustration of heparin sodium enhancing one-pot detection with various Cas12 proteins. g Characterization of the SURVEY method for targeting f3l or b6r when LbCas12a was substituted with AsCas12a. h Characterization of the SURVEY method for targeting f3l or b6r when LbCas12a was substituted with AapCas12b. Data are shown as mean ± s.d. for n = 3 biologically independent samples. Statistical significance was analyzed using a two-tailed t test: ns, p > 0.05. The schematics shown in Fig. 4f were created by figdraw.com.
Beyond LbCas12a, AsCas12a and AapCas12b are also widely used in CRISPR-Dx methods25,37. Unlike LbCas12a, AsCas12a and AapCas12b target the canonical PAMs sequence TTN (where N represents A, G, C, or T)38,39. We tried to develop the RPA-AsCas12a and RPA-AapCas12b one-pot systems for detecting f3l and b6r genes at 1 fM (Fig. 4f). In the system using RPA-AsCas12a, fluorescence was observed in the presence of 40 μg/mL heparin sodium only, indicating its capability to detect 1 fM of the f3l and b6r genes (Fig. 4g and Supplementary Fig. 25a). Interestingly, RPA-AapCas12b could detect 1 fM of the f3l and b6r genes without heparin sodium (Fig. 4h and Supplementary Fig. 25b). However, dosing 20 μg/mL heparin sodium not only accelerated the detection rate of RPA-AapCas12b, but also resulted in higher fluorescence (Fig. 4h and Supplementary Fig. 25b). These findings suggest that the one-pot methods developed with dosing heparin sodium were also applicable to other subtypes of Cas12.
Overall, the detection process based on heparin sodium required 15–20 min only, greatly reducing the detection time. Initially, we applied pseudoviruses of the mpox to validate the detection performance of SURVEY. These pseudoviruses were created by integrating the f3l and b6r genes into the adenovirus genome. Subsequently, we introduced multiple pathogens such as Mycoplasma pneumoniae, SARS-CoV-2, influenza A virus, adenovirus without f3l and b6r genes, Pseudomonas aeruginosa, and Helicobacter pylori to the detection system to verify the specificity of the SURVEY method. The results show that significant fluorescence signals were observed only in the presence of pseudovirus carrying the f3l or b6r genes, thus confirming the high specificity of the SURVEY method (Fig. 5a, b). To more comprehensively assess the specificity and sensitivity of the SURVEY method, we utilized pseudovirus carrying the f3l or b6r genes, randomly introducing them into saliva samples at various concentrations for nucleic acid extraction and subsequent detection. In the analysis of 27 samples, which included both positive samples with cycle threshold (Ct) values ranging from 24 to 36 and negative samples, the SURVEY method, when targeting the f3l gene, demonstrated 100% sensitivity and specificity, as compared to qPCR (Fig. 5c, e). When targeting the b6r gene, SURVEY achieved a sensitivity of 95.7% and maintained 100% specificity (Fig. 5d, f). These results demonstrate the high specificity and sensitivity of the SURVEY method for pathogens detection.

a, b Specific characterization of the SURVEY method using environmental pathogens such as SARS-CoV-2, adenovirus, influenza A virus, Mycoplasma pneumoniae, Pseudomonas aeruginosa, and Helicobacter pylori as targets (crRNA targeting f3l of mpox (a), and crRNA targeting b6r of mpox (b)).c, d Detection of various concentrations of pseudovirus (carrying f3l (c) or b6r (d) genes) introduced into saliva using both qPCR and the SURVEY method. The threshold was set as the negative control plus three times its standard deviation. Fluorescence values exceeding this threshold were defined as positive, while those below were defined as negative. e, f Sensitivity (also known as the true positive rate) defined as: Sensitivity = True Positives/(True Positives + False Negatives). Specificity (also known as the true negative rate) defined as: Specificity = True Negatives/(True Negatives + False Positives). qPCR serves as the gold standard for determining negative and positive results. Data are shown as mean ± s.d. for n = 3 biologically independent samples.
Rapid detection of influenza A and SARS-CoV-2 using the SURVEY
To further explore the practical application potential of the SURVEY method in actual scenarios, we selected influenza A virus as the detection target and designed crRNA targeting its matrix protein 2 (M2) encoding gene. The canonical PAM sequence of this gene recognized by the crRNA was TTTG. Initially, we evaluated the RPA-Cas12a one-pot system’s ability to detect synthetic DNA sequences encoding the M2 protein gene without heparin sodium. The system exhibited limited capability in detecting trace nucleic acids (Fig. 6a and Supplementary Fig. 26a). However, after the addition of heparin sodium, the LoD was reduced to 1 aM, comparable to qPCR (Fig. 6b, c and Supplementary Fig. 26b). Considering that the genome of influenza A virus is RNA, we used the DNA encoding the M2 protein as a template, transcribed it into RNA, and then conducted detection. By constructing the RT (Reverse Transcription)-RPA-Cas12a detection system, the performance of this one-pot assay was assessed. First, we recognized that incorporating a reverse transcription step required adjusting the heparin sodium concentration to regulate the cleavage activity of LbCas12a, thereby optimizing detection efficiency. Therefore, we evaluated the impact of different concentrations of heparin sodium on the RT-RPA-Cas12a one-pot detection system, finding that 5-10 μg/mL heparin sodium was appropriate (Supplementary Fig. 27a, b). Subsequent testing reveals that no fluorescence signal was yielded in the absence of heparin sodium (Fig. 6d and Supplementary Fig. 28a). In presence of heparin sodium, fluorescence signal was detected and the LoD for the RT-RPA-Cas12a detection system reached 1 aM (≈0.6 copies/μL, volume=30 μL), matching the LoD of RT-qPCR (Fig. 6e, f and Supplementary Fig. 28b). Subsequently, we also introduced multiple pathogens such as Mycoplasma pneumoniae, SARS-CoV-2, lentivirus, adenovirus, Pseudomonas aeruginosa, and Helicobacter pylori to the detection system to verify the specificity of the SURVEY method. The results show that significant fluorescence signals were observed only in the presence of influenza A virus, thus confirming the high specificity of the SURVEY method (Supplementary Fig. 28c). Furthermore, the SURVEY method was used to detect 33 clinical samples, successfully identifying influenza A in 21 samples (Fig. 6g, h). Moreover, the SURVEY successfully identified all negative samples (Fig. 6h). Compared to RT-qPCR, its specificity was 100% and the sensitivity was 95.5% (Fig. 6i).

a–c LoD analysis for the synthetic M2 protein coding gene (DNA) using the RPA-Cas12a one-pot detection method without (a) and with (b) 40 µg/mL heparin sodium, and qPCR (c) at target concentrations of 0, 0.1, 1, 10, 100, 1000, 10000 aM. d–f LoD analysis for transcribed mRNA targets using the RT-RPA-Cas12a one-pot detection method without (d) and with (e) 10 µg/mL heparin sodium, and RT-qPCR (f) at target concentrations of 0, 0.1, 1, 10, 100, 1000, 10000 aM. g Schematic representation of detection of human saliva and sputum samples. h Analysis of samples from all 33 patients using RT-qPCR and the SURVEY method. RT-qPCR was performed for 40 cycles. For SURVEY detection, the threshold was set to the negative control fluorescence value plus three times its standard deviation; samples exceeding this threshold were classified as positive, while those below were considered negative. i Specificity and sensitivity analysis for samples detection using the SURVEY method, with RT-qPCR serving as the gold standard for determining negative and positive results. Data are shown as mean ± s.d. for n = 3 biologically independent samples. Statistical significance was analyzed using a two-tailed t test: ns, p > 0.05. The schematics shown in Fig. 6g were created by figdraw.com.
Beyond human samples, we extended to pathogen detection in wastewater40. Since the COVID-19 pandemic began, wastewater-based epidemiology (WBE) has emerged as a pivotal tool because it captures the infection dynamics within catchment area, facilitating early warnings and detailed analyses of epidemic spread41,42. The rapid development of WBE underscores the necessity for molecular diagnostic methods capable of on-site detection, which can provide in-situ alerts about emerging outbreaks43,44. In-situ detection methods conducted at wastewater sites must possess rapidity, high sensitivity, and specificity, as well as an absence of nucleic acid aerosol contamination45. Therefore, we evaluated the performance of the SURVEY method for the detection of SARS-CoV-2 in wastewater. By targeting the gene encoding the N (nucleocapsid protein) of SARS-CoV-246, we initially tested the LoD of the SURVEY method on synthetic DNA alone. Similar to the results with mpox pseudovirus and influenza A virus, a LoD of 1 aM (≈0.6 copies/μL, volume=30 μL) was achieved only in the presence of heparin sodium (Fig. 7a–c and Supplementary Fig. 29a, b). Subsequently, we established an RT-RPA-Cas12a one-pot detection method for SARS-CoV-2 RNA. Detection results show that the LoD of the SURVEY method was also 1 aM, equal to that of RT-qPCR (Fig. 7d–f and Supplementary Fig. 30a, b). Similar cross-reactivity tests further confirmed the high specificity of SURVEY targeting SARS-CoV-2 (Supplementary Fig. 30c).

a–c LoD analysis for the synthetic N protein coding gene (DNA) using the RPA-Cas12a one-pot detection method without (a) and with (b) 40 µg/mL heparin sodium, and qPCR (c) at target concentrations of 0, 0.1, 1, 10, 100, 1000, 10000 aM. Data are shown as mean ± s.d. for n = 3 biologically independent samples. d–f LoD analysis for transcribed mRNA targets using the RT-RPA-Cas12a one-pot detection method without (d) and with (e) 10 µg/mL heparin sodium, and RT-qPCR (f) at target concentrations of 0, 0.1, 1, 10, 100, 1000, 10000 aM. Data are shown as mean ± s.d. for n = 3 biologically independent samples. g Schematic representation of SARS-CoV-2 detection in wastewater samples from 10 provinces in China. h Analysis of samples from all 25 influents and 25 effluents using RT-qPCR and the SURVEY method. RT-qPCR was performed for 40 cycles. For SURVEY detection, the threshold was set to the negative control fluorescence value plus three times its standard deviation; samples exceeding this threshold were classified as positive, while those below were considered negative. Statistical significance was analyzed using a two-tailed t test: ns, p > 0.05. The schematics shown in (g) were created by figdraw.com.
During the peak of the COVID-19 pandemic, we collected influent and effluent samples from 25 wastewater treatment plants (WWTPs) across 10 cities in China on December 26, 2022 (Fig. 7g). Viruses were concentrated from these samples using polyethylene glycol precipitation47. We employed both RT-qPCR and the SURVEY method to detect viruses in these wastewater samples. The findings indicate that SARS-CoV-2 was detectable in the influent of all 25 WWTPs, suggesting widespread transmission of the virus across China at that time (Fig. 7h). However, SARS-CoV-2 was not detected in the effluent from any WWTPs, indicating that the activated sludge process along with filtration and disinfection measures was effective in removing the viruses from the municipal wastewater (Fig. 7h). Additionally, when comparing the SURVEY method with RT-qPCR, SURVEY identified 24 out of 25 positive samples and accurately recognized all negative samples, achieving a sensitivity of 96% and specificity of 100% (Supplementary Fig. 31a, b).
In conclusion, leveraging the inhibitory effects of heparin sodium on Cas12a’s cleavage activity, we developed a versatile one-pot detection strategy. Demonstrating high sensitivity and specificity, this approach was effective across both canonical and suboptimal PAMs, enabling rapid detection (15–20 min) of pathogens such as mpox pseudovirus, influenza A virus, and SARS-CoV-2. This method offers a valuable advancement in rapid diagnostic solutions for a range of infectious diseases and has potential implications for enhancing in-situ diagnostics in clinical settings and environmental monitoring, particularly with human and wastewater samples.
Discussion
The large-scale epidemics such as COVID-19 has significantly accelerated the development of POCT processes, highlighting the potential of CRISPR-Dx technologies for on-site rapid diagnostics to both the scientific community and the public48,49. A critical step in advancing CRISPR-Dx technologies integrated with isothermal amplification towards POCT is the development of one-pot detection methods6. The core scientific challenge for one-pot detection is how to precisely regulating the cleavage activity of Cas1223,28. Previous studies into the cleavage activity of Cas12 lacked precise methods for controlling cleavage activity, prompting the exploration of alternative strategies such as spatial isolation, high-viscosity glycerol media, or light-controlled crRNA to facilitate one-pot detection29,32,50. In this work, we discover that the cleavage activity of Cas12a could be regulated by varying heparin sodium concentration via interference with the Cas12a-crRNA binding process. Leveraging this characteristic of heparin sodium, we develop the (RT)-RPA-Cas12a-based one-pot detection technology-SURVEY, which not only extends the applicability of PAM sequences, but also substantially reduces the detection time. Importantly, the inhibitory effect of heparin sodium enables one-pot detection using both AsCas12a and AapCas12b, thereby expanding the applicability scope of SURVEY. Our method demonstrates marked advantages over previous technologies in terms of LoD, detection speed, PAMs selection, and subtypes of Cas12 (Table 1). Moreover, each assay requires a minimal amount of heparin sodium (10-40 μg/mL) only at per thousand cost of approximately $0.01 to $0.04, rendering the expense virtually negligible (Supplementary Table 6).
Heparin sodium is primarily used as an anticoagulant in clinical settings, especially in surgical procedures51. Despite an emerging report on the inhibition of Cas12a cleavage activity by heparin sodium during the course of our manuscript preparation, that study did not explore the incorporation of heparin sodium into CRISPR-Dx52. Our study reports the use of heparin sodium in CRISPR-Dx systems for trace nucleic acid detection, pioneering potential applications for this substance. We find that heparin sodium primarily interferes with the binding between Cas12a and crRNA. Our MD simulations elucidate the dynamic process of competition between heparin and crRNA for Cas12a, while further exploration is necessary to identify the key amino acid residues that form stable interactions with heparin sodium. In the RPA-Cas12a system, dosing heparin sodium inhibits Cas12a’s cleavage of trace substrates to enables RPA to perform extensive nucleic acid amplification. When the nucleic acid amplification products reach a certain concentration, Cas12a’s cleavage activity is no longer effectively inhibited, thereby generating significant fluorescence signals. Notably, in our study, the required concentration of heparin sodium varied depending on the buffer used. Therefore, in practical applications, it is essential to optimize the concentration of heparin sodium during the preliminary experiments to develop the most efficient one-pot detection system. Additionally, it is expected that heparin sodium may also regulate the cleavage activities of other Cas proteins such as Cas9 and Cas13. By elucidating the molecular mechanisms of heparin sodium’s effects on Cas9, Cas12, and Cas13, potential applications of heparin sodium in regulating intracellular gene editing processes could be expanded to address issues of immune evasion caused by high cleavage activity53.
Heparin sodium-mediated one-pot detection technology has resolved the problems related to nucleic acid aerosol contamination, sensitivity, and PAM limitations. For the human-derived samples, the qualitative results provided by CRISPR-Dx can meet the general needs of clinical and household testing. However, for environmental samples, such as wastewater and activated sludge, precise abundance data are often required to indicate pathogen levels and infection trends in populations. The CRISPR-Dx technology, combined with isothermal amplification, struggles to achieve accurate quantification. Therefore, there is an urgent need to focus on the development of quantitative CRISPR-Dx technology in the future. Ultimately, a pivotal challenge in limiting the spread of CRISPR-Dx technology lies in reducing the overall cost of testing to levels comparable with antigen testing, particularly the costs associated with RPA reagents and Cas proteins. Future CRISPR-Dx studies should focus more on real-clinical and environmental applications and the corresponding engineering challenges to further advance the development of POCT.
Methods
Ethical statement
All experiments involving saliva and sputum samples were approved by the Ethics Committee of the Fujian Medical University Union Hospital, China (2024KY093). All patients provided informed written consent. No participant compensation was provided.
Plasmid, Monkeypox pseudovirus and DNA/RNA standards preparation
The target plasmids containing the monkeypox virus genes f3l and b6r (mpox_USA_2022_MA001, accession: ON563414), SARS-CoV-2 nucleocapsid protein (accession: MN975262.1), or the influenza A virus M2 protein encoding genes (NC_002016.1) were synthesized by General Bio Co., China. Replication-deficient Adenovirus type 5 (AD5) containing the f3l or b6r genes of mpox was provided by Sangon Biotech Co., China. The pseudoviruses were diluted with a 1:100 ratio in lysis buffer (total nucleic acid release agent, Genestonebio Co., China) to prepare mpox pseudovirus samples. Plasmids containing the aforementioned four genes were extracted and quantified, followed by serial dilution and detection using both qPCR and SURVEY. Transcripts of the N and M2 proteins encoding gene were prepared by using T7 promoter, serially diluted to standard concentrations, and detected using RT-qPCR and SURVEY.
Expression and purification of LbCas12a
The overexpression plasmid pET28a-LbCas12a was transformed into Escherichia coli BL21 Rosetta (DE3). After overnight cultivation in 5 mL of Luria-Bertani (LB) medium, the strains were transferred to 3 L of fresh LB medium for further cultivation. The culture was incubated at 37 °C and 220 rpm until the OD600 reached 0.8. Then, 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added as an inducer, and incubation continued at 16 °C for 16 h. Cells were collected by centrifugation and subsequently resuspended in buffer containing 50 mM Tris-HCl (pH 8.0) and 1.5 M NaCl for ultrasonication. The supernatant was purified using Ni-NTA agarose chromatography. The concentration of LbCas12a protein was determined using a modified Bradford protein assay kit (Sangon Bio Co., China) based on bovine serum albumin and stored at -80 °C for future use. AsCas12a and AapCas12b were purchased from Tolobio Co., China.
Preparation of crRNA
To prepare the transcription template of crRNA, two oligonucleotides were first heated at 95 °C for 4 min and then cooled to 4 °C at a rate of 0.1 °C/s. One of the oligonucleotides included the T7 promoter sequence, while the other contained a handle and spacer sequence. These templates were incubated with T7 RNA polymerase at 37 °C for 2 h to synthesize the target crRNA. The transcription products were purified by digesting the DNA template with DNase I at 37 °C and removing impurities using an RNA purification kit (Jianshi Biotechnology Co., China). All crRNA spacer sequences are listed in Supplementary data 1.
Characterizations of cis and trans cleavage activities
In the activity characterization tests, heparin sodium at various concentrations (0, 0.1, 0.2, 0.3, 0.4, 0.5, and 2.0 µg/mL) was added to a solution containing 50 nM LbCas12a and 200 nM crRNA in Buffer-1. For the cis-cleavage experiments, 10 ng/µL of plasmid or DNA fragments was added to the mixture, reacted at 37 °C for 30 min, and then treated at 80 °C for 20 min, followed electrophoresis on a 1% agarose gel. For the fluorescence characterization of cis-cleavage, 500 nM of pre-annealed DNA targets labeled with FAM and BHQ1 in a 10:1 ratio were added to the reaction system, and the fluorescence generated post-cleavage was measured using a SpectraMax i3x multimode microplate reader. For the fluorescence characterization of trans-cleavage, 10 nM of pre-annealed DNA target unlabeled without FAM and BHQ1 and a single-stranded DNA probe labeled with FAM and BHQ1 were introduced to the system for fluorescence detection. All probe sequences are listed in Supplementary Table 1.
Electrophoretic mobility shift assay
To assess the binding capability of LbCas12a to crRNA, a mixture containing 1.2 µM LbCas12a and 1.2 µM crRNA was prepared. The control group contained 0 µg/mL heparin sodium, while the experimental group included 600 µg/mL of heparin sodium. The mixtures were run on a PAGE gel at 140 V for 50 min, followed by staining in 1×TAE solution containing nucleic acid stain. When Cas12a bound to crRNA, the complex remained at the top of the lane, while unbound crRNA was imaged at the bottom of the lane.
MD simulation
MD simulations were based on cryo-EM structure of LbCas12a (PDB: 6P7M) at 3.00 Å resolution. Missing residues in the 6P7M were reconstructed through Alphafold3 webserver (https://golgi.sandbox.google.com/). The structure of Alphafold3-predicted Cas12a-crRNA complex was highly similar to that of 6P7M as indicated by the low RMSD value between the two structures (Supplementary Fig. 6 c,d). Hence, the predicted Cas12a was utilized as the initial structure for MD simulation. Due to the close molecular weight as the crRNA (Supplementary Fig. 7a,b), heparin dp24 (PDB: 3irj) was selected and further modified to remove poor contacts for MD simulation. To investigate the impact of negative charge on the interaction of heparin with Cas12a, sulfur-free heparin (S-free heparin) model was constructed by deleting the -SO3H groups of heparin and sealing the exposed N and O with H (Supplementary Fig. 7c). The Amber ff99SB-ILDN force field was employed to parameterize the Cas12a/crRNA molecules54. The GAFF force field parameters for heparin and S-free heparin were generated by Sobtop55. All systems were solvated in a cubic box with TIP3P water molecules and counterions were added to neutralize the systems. To match the activity characterization experiments, the crRNA was moved out of the initial binding site and placed at a centroid distance of > 5 nm away from Cas12a. For comparison, various binary and ternary systems were constructed, including Cas12a-heparin, Cas12a-S-free-heparin, Cas12a-crRNA-heparin and Cas12a-crRNA-S-free-heparin (Supplementary Fig. 9).
MD simulations were performed using the GROMACS 2022 package56. The systems were subjected to energy minimization, 500 ps NVT equilibration, 500 ps NPT equilibration, and 100 ns production MD steps. To remove steric clashes and optimize the systems, each minimization was composed of the steepest decent algorithm with a tolerance of 500.0 kJ/mol/nm followed by the conjugate gradient step with a lower tolerance of 100.0 kJ/mol/nm. The NVT equilibration was performed using the md integrator with a time step of 2 fs. The temperature was linearly increased from 0 K to 298.15 K in 100 ps and was maintained at 298.15 K using the V-rescale thermostat with a time constant of 0.2 ps. The NPT equilibration was performed using the same setups as the NVT ensemble with the addition of a Parrinello-Rahman barostat with an isotropic pressure coupling type. During these two equilibrations, bonds including hydrogens were constrained using the LINCS algorithm and a positional restraint was applied to all solute heavy atoms. Over the course of 100 ns MD simulations, the restraints on the heavy atoms were canceled and the md integrator with a time step of 2 fs was adopted. The temperature was maintained at 298.15 K for both the protein-ligand group and the environment group using the V-rescale thermostat with a time constant of 0.2 ps. The system was coupled to a Parrinello-Rahman barostat with an isotropic pressure coupling type. The pressure was maintained at 1.0 bar with a time constant of 2.0 ps and a compressibility of 4.5e-5 bar-1. The hydrogen bonds were constrained using the LINCS algorithm. Electrostatic interactions were calculated using the PME algorithm with a cutoff radius of 1.0 nm. Van der Waals interactions were calculated using the cut-off algorithm with a cutoff radius of 1.0 nm.
Post-simulation analysis was conducted using GROMACS tools to assess the stability and conformational changes of the Cas12a-crRNA/heparin complex. Visualization of the MD trajectories was performed using VMD and UCSF ChimeraX, enabling detailed examination of the molecular interactions and dynamics. The binding free energy for the association of crRNA and heparin with Cas12a was calculated using the Molecular Mechanics with Generalized Born and Surface Area (MM-GBSA) method57.
The trans-cleavage activities of Cas12 with chondroitin sulfate or buffers
The inhibitory effects of chondroitin sulfate at various concentrations (0, 25, 50, 100, 200, and 300 µg/mL) (Sangon Bio Co., China) were examined. The assay for measuring nuclease activity in the presence of chondroitin sulfate was consistent with that used for heparin sodium, employing fluorescence intensity to quantify the inhibition of nuclease activity by these compounds. In the characterization of cleavage activity, three different buffers, labeled as Buffer-1, Buffer-2, Buffer-3 and RPA buffers were utilized. Buffer-1 consisted of 40 mM HEPES, 100 mM NaCl, and 20 mM MgCl2 (pH 7.4). Buffer-2 was composed of 6.5% polyethylene glycol, 100 mM HEPES, 20 mM magnesium acetate, and 1.5% mannitol (pH 7.4). Buffer-3 was the commercially available Cut Smart buffer from NEB. The RPA buffer is prepared by dissolving lyophilized particles in 29.4 µL of Buffer A and adding 6 µL of Buffer B to form a mixture. In the inhibition experiments, the total reaction volume was 20 µL, containing 4 µL of Buffer-1, 10 µL of Buffer-2, 2 µL of Buffer-3 or 10 µL RPA buffer.
qPCR and RT-qPCR detections
The qPCR detection of samples containing mpox plasmid standards was carried out in a 20 µL reaction system, consisting of 10 µL of 2×AceQ qPCR Probe Master Mix (Vazyme Biotech Co., China), 1 µL of each primer pair (10 µM), and 0.2 µL of 10 µM TaqMan probe (Sangon Biotech Co., China). RT-qPCR detection for influenza virus A and SARS-CoV-2 samples was similarly performed in a 20 µL reaction system, including 10 µL of 2×AceQ qPCR Probe Master Mix, 0.5 µL reverse transcriptase (Takara Biotech Co., China), 1 µL of each primer pair (10 µM), and 0.2 µL of 10 µM TaqMan probe. Details of the primers and TaqMan probes are listed in Supplementary Table 2.
One-pot detection
In the one-pot detection experiments, the impact of heparin sodium on nuclease activity using different concentrations of plasmid standards with traditional or suboptimal PAMs was determined. Lyophilized RPA particles (Weifang Amp-Future Biotech Co., China), were reconstituted in the reaction mixture containing 29.4 µL of Buffer A, 2 µL each of 20 µM RPA forward and reverse primers, 400 nM FQ-ssDNA reporter molecule, 100 nM LbCas12a (AsCas12a, AapCas12b), 200 nM crRNA, and nuclease-free water, along with various concentrations of heparin sodium. After adding 3 µL of Buffer B, the 24 µL mixture was incubated at 37 °C. Fluorescence was measured using a SpectraMax i3x multimode microplate reader at an excitation wavelength of 485 nm and emission wavelength of 520 nm. To investigate the effect of different crRNA spacer lengths on the efficiency of a one-pot detection method, the f3l gene with a target concentration of 1 fM was selected. The spacer lengths were set to 10, 15, 17-20, 25, and 30 nt, and the heparin sodium concentrations were 0 or 40 µg/mL. The reaction was conducted at 37 °C for 20 min. The fluorescence values were compared to assess the impact of different spacer lengths and heparin sodium concentrations. For processing influenza A and SARS-CoV-2 samples, the reaction additionally included 8 µL of 20 µM reverse transcription primer and 0.8 µL of reverse transcriptase (HiScript III reverse transcriptase, Vazyme Biotech Co., China). Subsequently, the RT-RPA-CRISPR system was incubated at 37 °C for 1 h, and fluorescence was measured using a SpectraMax i3x multimode microplate reader. Sequences of primers and probes are detailed in Supplementary Table 2.
Sample collection and processing
To validate the specificity of SURVEY, interference samples including SARS-CoV-2 (Fujian Medical University Union Hospital, China), lentivirus (Sangon Bio Co., China), adenovirus (Sangon Bio Co., China), influenza A virus (Fujian Medical University Union Hospital, China), Mycoplasma pneumoniae (Fujian Medical University Union Hospital, China), Helicobacter pylori (Guangdong Microbial Culture Collection Center, China), and Pseudomonas aeruginosa PAO1 (Laboratory storage) were collected. Influenza A virus samples included saliva and sputum samples from 33 patients at the Fujian Medical University Union Hospital, China. This study did not consider sex and gender. The sex and gender of participants was determined based on self-report. Detailed information on patient gender, age, quantity, and RT-qPCR results are provided in Supplementary Table 3. SARS-CoV-2 samples were collected from influents and effluents at 25 WWTPs across 10 provinces in China on December 26, 2022. Details of the selected WWTPs are listed in Supplementary Tables 4-5. Samples were stored at 0 to 4 °C and transported to the laboratory for analysis within 48 h. All patient samples were initially inactivated at 65 °C for 2 h, followed by nucleic acid extraction and purification using an RNA extraction kit. The obtained RNA were purified using one-step PCR inhibitor removal columns (Genstone Biotech Co., China). For wastewater samples, after a similar inactivation process, viruses were concentrated using the reported polyethylene glycol precipitation method47. The nucleic acids were subsequently extracted and purified. The extracted nucleic acids were stored at -80 °C until analysis.
Statistics and reproducibility
Statistical analysis was performed using Microsoft Excel (2016) and Origin Pro (Version 9.5.0 SR1). Significance testing was conducted using a two-tailed t-test, with a significance level set at P < 0.05. Randomized sampling was employed for the analysis of all mpox pseudovirus, influenza A virus, and SARS-CoV-2 samples. All experimental results were derived from at least three biologically independent experiments, and data are presented as the mean ± standard deviation (mean ± s.d.).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are provided in the Source Data File. The protein structure used in this work includes the PDB accession code 6P7M. Any additional inquiries should be addressed to Dong-Feng Liu or Han-Qing Yu (corresponding author). Source data are provided with this paper.
References
Pacheco, L. D., Weaver, S. C. & Saade, G. R. Zika virus Infection—after the Pandemic. N. Engl. J. Med. 382, E3 (2020).
Piot, P., Muyembe, J.-J. & Edmunds, W. J. Ebola in west Africa: from disease outbreak to humanitarian crisis. Lancet Infect. Dis. 14, 1034–1035 (2014).
Broughton, J. P. et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020).
Jiang, T. et al. Creating an ultra-sensitive detection platform for monkeypox virus DNA based on CRISPR technology. J. Med. Virol. 95, e29121 (2023).
Collaborators, G. B. D. D. Global age-sex-specific mortality, life expectancy, and population estimates in 204 countries and territories and 811 subnational locations, 1950-2021, and the impact of the COVID-19 pandemic: a comprehensive demographic analysis for the Global Burden of Disease Study 2021. Lancet 403, 1989–2056 (2024).
Tong, X. et al. Fast and sensitive CRISPR detection by minimized interference of target amplification. Nat. Chem. Biol. https://doi.org/10.1038/s41589-023-01534-9 (2024).
Osorio, N. S. & Correia-Neves, M. Implication of SARS-CoV-2 evolution in the sensitivity of RT-qPCR diagnostic assays. Lancet Infect. Dis. 21, 166–167 (2021).
Blairon, L., Piteus, S., Beukinga, I. & Tre-Hardy, M. Development and implementation of a RT-qPCR extraction-free protocol for the detection of SARS-CoV-2 and impact on the turn-around-time. J. Med. Virol. 93, 2538–2542 (2021).
Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 5, 1299–1305 (2020).
Rabe, B. A. & Cepko, C. SARS-CoV-2 detection using isothermal amplification and a rapid, inexpensive protocol for sample inactivation and purification. Proc. Natl Acad. Sci. USA 117, 24450–24458 (2020).
Zhang, G., Brown, E. W. & Gonzalez-Escalona, N. Comparison of real-time PCR, reverse transcriptase real-time PCR, loop-mediated isothermal amplification, and the FDA conventional microbiological method for the detection of Salmonella spp. in produce. Appl. Environ. Microbiol. 77, 6495–6501 (2011).
Du, Z., Tian, L. & Jin, D. Y. Understanding the impact of rapid antigen tests on SARS-CoV-2 transmission in the fifth wave of COVID-19 in Hong Kong in early 2022. Emerg. Microbes Infect. 11, 1394–1401 (2022).
Mak, G. C. K. et al. Evaluation of rapid antigen test for detection of SARS-CoV−2 virus. J. Clin. Virol. 129, 104500 (2020).
Cherkaoui, D., Huang, D., Miller, B. S., Turbe, V. & McKendry, R. A. Harnessing recombinase polymerase amplification for rapid multi-gene detection of SARS-CoV−2 in resource-limited settings. Biosens. Bioelectron. 189, 113328 (2021).
Saki, E. F., Setiawan, S. A. & Wicaksono, D. H. B. Portable tools for COVID-19 point-of-care detection: a review. Ieee Sens. J. 21, 23737–23750 (2021).
Gootenberg, J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–438 (2018).
Bravo, J. P. K. et al. RNA targeting unleashes indiscriminate nuclease activity of CRISPR-Cas12a2. Nature 613, 582–587 (2023).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Chen, J. et al. Trans-nuclease activity of Cas9 activated by DNA or RNA target binding. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02255-7 (2024).
Li, S. Y. et al. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell. Res. 28, 491–493 (2018).
Li, L. et al. HOLMESv2: a CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. Acs. Synth. Bio. 8, 2228–2237 (2019).
Lu, S. et al. Fast and sensitive detection of SARS-CoV-2 RNA using suboptimal protospacer adjacent motifs for Cas12a. Nat. Biomed. Eng. 6, 286–297 (2022).
Yan, H. et al. A one-pot isothermal Cas12-based assay for the sensitive detection of microRNAs. Nat. Biomed. Eng. 7, 1583–1601 (2023).
Yang, L. et al. A PAM-free one-step asymmetric RPA and CRISPR/Cas12b combined assay (OAR-CRISPR) for rapid and ultrasensitive DNA detection. Anal. Chem. 96, 5471–5477 (2024).
Li, S. Y. et al. CRISPR-Cas12a-assisted nucleic acid detection. Cell. Discov. 4, 20 (2018).
Hu, M. et al. Light-start CRISPR-Cas12a reaction with caged crRNA enables rapid and sensitive nucleic acid detection. Angew. Chem.-Int. Ed. 62, e202300663 (2023).
Hu, M. et al. Photocontrolled crRNA activation enables robust CRISPR-Cas12a diagnostics. Proc. Natl Acad. Sci. USA 119, e2202034119 (2022).
Lin, M. et al. Glycerol additive boosts 100-fold sensitivity enhancement for one-pot RPA-CRISPR/Cas12a assay. Anal. Chem. 94, 8277–8284 (2022).
Yin, K. et al. Dynamic aqueous multiphase reaction system for one-pot CRISPR-Cas12a-based ultrasensitive and quantitative molecular diagnosis. Anal. Chem. 92, 8561–8568 (2020).
Wang, B. et al. Cas12aVDet: a CRISPR/Cas12a-based platform for rapid and visual nucleic acid detection. Anal. Chem. 91, 12156–12161 (2019).
Chen, Y. et al. Photoactivatable CRISPR/Cas12a strategy for one-pot DETECTR molecular diagnosis. Anal. Chem. 94, 9724–9731 (2022).
Cheng, Z.-H. et al. Optimized antibiotic resistance genes monitoring scenarios promote sustainability of urban water cycle. Environ. Sci. Technol. 58, 9636–9645 (2024).
Wang, X. et al. Rapid and sensitive Cas13a/Cas12a-based one-pot dual-target strategy to detect monkeypox virus and its co-infected viruses. Sci. Bull. 68, 3142–3148 (2023).
Palermo, G., Miao, Y., Walker, R. C., Jinek, M. & McCammon, J. A. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl Acad. Sci. USA 114, E8944 (2017).
Yamano, T. et al. Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol. Cell 67, 633–645 (2017).
Cao, Y. et al. CoLAMP: CRISPR-based one-pot loop-mediated isothermal amplification enables at-home diagnosis of SARS-CoV-2 RNA with nearly eliminated contamination utilizing amplicons depletion strategy. Biosens. Bioelectron. 236, 115402 (2023).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Nguyen, L. T. et al. A thermostable Cas12b from Brevibacillus leverages one-pot discrimination of SARS-CoV-2 variants of concern. Ebiomedicine 77, 103926 (2022).
Ahmed, F. et al. Wastewater-based monitoring could help guide responses to the USA opioid epidemic. Nat. Water 1, 401–404 (2023).
Li, X. et al. Wastewater-based epidemiology predicts COVID-19-induced weekly new hospital admissions in over 150 USA counties. Nat. Commun. 14, 4548 14 (2023).
Singer, A. C. et al. A world of wastewater-based epidemiology. Nat. Water 1, 408–415 (2023).
Cao, H. et al. Paper device combining CRISPR/Cas12a and reverse-transcription loop-mediated isothermal amplification for SARS-CoV-2 detection in wastewater. Environ. Sci. Technol. 56, 13245–13253 (2022).
Yang, Y., Wang, F., Xue, B. & Zhou, X. Field-deployable assay based on CRISPR-Cas13a coupled with RT-RPA in one tube for the detection of SARS-CoV-2 in wastewater. J. Hazard. Mater. 459, 132077 (2023).
Yang, Z. Low-cost and rapid sensors for wastewater surveillance at low-resource settings. Nat. Water 1, 405–407 (2023).
Xu, X. et al. Evaluation of RT-qPCR primer-probe sets to inform public health interventions based on COVID-19 sewage tests. Environ. Sci. Technol. 56, 8875–8884 (2022).
Zheng, X. et al. A rapid, high-throughput, and sensitive PEG-precipitation method for SARS-CoV-2 wastewater surveillance. Water Res. 230, 119560 (2023).
Tian, T. et al. An ultralocalized Cas13a assay enables universal and nucleic acid amplification-free single-molecule RNA diagnostics. ACS Nano 15, 1167–1178 (2021).
Arizti-Sanz, J. et al. Simplified Cas13-based assays for the fast identification of SARS-CoV-2 and its variants. Nat. Biomed. Eng. 6, 932–943 (2022).
Wei, J. et al. MASTR Pouch: Palm-size lab for point-of-care detection of Mpox using recombinase polymerase amplification and CRISPR technology. Sens. Actuators B: Chem. 390, 133950 (2023).
Wang, P. et al. Heparin: an old drug for new clinical applications. Carbohydr. Polym. 295, 119818 (2022).
Cao, M. et al. Heparin specifically inhibits CRISPR/Cas12 activation, enabling ultrasensitive heparin detection and gene editing regulation. Anal. Chem. 96, 3970–3978 (2024).
Collias, D. et al. Systematically attenuating DNA targeting enables CRISPR-driven editing in bacteria. Nat. Commun. 14, 680 (2023).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins-Struct. Funct. Bioinform. 78, 1950–1958 (2010).
LU, T. Sobtop, Version 1.0[EB/OL]. http://sobereva.com/soft/sobtop (2022).
Paul Bauer, B. H., & Erik Lindahl. GROMACS 2022 Manual. https://doi.org/10.5281/zenodo.6103568 (2022).
Valdes-Tresanco, M. S., Valdes-Tresanco, M. E., Valiente, P. A. & Moreno, E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 17, 6281–6291 (2021).
Wang, P. et al. One-pot molecular diagnosis of acute hepatopancreatic necrosis disease by recombinase polymerase amplification and CRISPR/Cas12a with specially designed crRNA. J. Agric. Food Chem. 71, 6490–6498 (2023).
Malcı, K., Walls, L. E. & Rios-Solis, L. Rational design of CRISPR/Cas12a-RPA based one-pot COVID-19 detection with design of experiments. ACS Synth. Biol. 11, 1555–1567 (2022).
de Puig, H. et al. Minimally instrumented SHERLOCK (miSHERLOCK) for CRISPR-based point-of-care diagnosis of SARS-CoV-2 and emerging variants. Sci. Adv. 7, eabh2944 (2021).
Tan, Q. et al. Simple, sensitive, and visual detection of 12 respiratory pathogens with one-pot-RPA-CRISPR/Cas12a assay. J. Med. Virol. 96, 29624 (2024).
Chen, Y., Mei, Y., Zhao, X. & Jiang, X. Reagents-loaded, automated assay that integrates recombinase-aided amplification and Cas12a nucleic acid detection for a point-of-care test. Analy. Chem. 92, 14846–14852 (2020).
Ding, X. et al. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 11, 4711 (2020).
Shao, F., Park, J. S., Zhao, G., Hsieh, K. & Wang, T.-H. Elucidating the role of CRISPR/Cas in single-step isothermal nucleic acid amplification testing assays. Anal. Chem. 95, 3873–3882 (2023).
Aman, R. et al. iSCAN-V2: a one-pot RT-RPA-CRISPR/Cas12b assay for point-of-care SARS-CoV-2 detection. Front. Bioeng. Biotechnol. 9, 800104 (2022).
Wang, X. E. et al. CDetection.v2: one-pot assay for the detection of SARS-CoV-2. Front. Microbiol. 14, 1158163 (2023).
Jing, W. et al. CHAMP: a centrifugal microfluidics-based CRISPR/Cas12b-combined real-time LAMP one-pot method for mycoplasma pneumoniae infection diagnosis. ACS Omega 9, 38989–38997 (2024).
Joung, J. et al. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N. Engl. J. Med. 383, 1492–1494 (2020).
Nguyen, L. T. et al. Engineering highly thermostable Cas12b via de novo structural analyses for one-pot detection of nucleic acids. Cell Rep. Med. 4, 101037 (2023).
Acknowledgements
The authors wish to thank the National Natural Science Foundation of China (52192684-H.-Q.Y, 51821006-H.-Q.Y, 52322002-D.-F.L. and 52400255-Z.-H.C.), the Major Science and Technology Projects of Anhui Province (202203a07020015-D.-F.L.), the Science Fund for Distinguished Young Scholars of Anhui Province (2208085J36-D.-F.L.), and the Postdoctoral Fellowship Program of CPSF under Grant Number GZC20241642-Z.-H.C. for supporting this work.
Author information
Authors and Affiliations
Contributions
D.F.L. and H.Q.Y. designed this work. Z.H.C., X.Y.L., S.S.Y., X.F.L., D.F.L., and H.Q.Y. performed the experimental investigations. Z.H.C., D.M., S.X.Z., J.J.C., and X.Y.L. performed the data analyses. All authors contributed to the interpretation of the findings. Z.H.C., X.Y.L., S.S.Y., D.F.L., and H.Q.Y. wrote and edited. All the authors contributed to discussion of the results and the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Piyush Jain, Xiaoming Zhou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, ZH., Luo, XY., Yu, SS. et al. Tunable control of Cas12 activity promotes universal and fast one-pot nucleic acid detection. Nat Commun 16, 1166 (2025). https://doi.org/10.1038/s41467-025-56516-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56516-3
