
Abstract
There are 90 independent genome-wide significant genetic risk variants for Parkinson’s disease (PD) but currently only five nominated loci for PD progression. The biology of PD progression is likely to be of central importance in defining mechanisms that can be used to develop new treatments. We studied 6766 PD patients, over 15,340 visits with a mean follow-up of between 4.2 and 15.7 years and carried out genome-wide survival studies for time to a motor progression endpoint, defined by reaching Hoehn and Yahr stage 3 or greater, and death (mortality). There was a robust effect of the APOE ε4 allele on mortality in PD. We also identified a locus within the TBXAS1 gene encoding thromboxane A synthase 1 associated with mortality in PD. We also report 4 independent loci associated with motor progression in or near MORN1, ASNS, PDE5A, and XPO1. Only the non-Gaucher disease causing GBA1 PD risk variant E326K, of the known PD risk variants, was associated with mortality in PD. Further work is needed to understand the links between these genomic variants and the underlying disease biology. However, these may represent new candidates for disease modification in PD.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative condition for which there are no drug treatments to stop or slow disease progression. Large-scale genome-wide case-control association studies (GWASs) of PD have identified 90 independent variants associated with disease risk1. However, it is also important to study the genetics and biology of disease progression. This will enable the development of potential disease-modifying treatments. There have now been a handful of GWAS that aim to identify genetic variants associated with progression in PD. These have nominated loci in SLC44A1 (encoding choline transporter-like protein -1, involved in membrane synthesis) for progression to Hoehn and Yahr (H&Y) stage 3 or greater2, APOE for cognitive progression3, LRP1B (encoding a low-density lipoprotein receptor which is involved in amyloid precursor protein trafficking) for progression to dementia4, and RIMS2 (encoding the RAB3 interacting RIMS2 protein, involved in neurotransmitter release) for progression to PD dementia5. In addition, many candidate gene studies have reported that variants in GBA1, APOE, and MAPT, are associated with the rate of PD motor and cognitive progression6.
PD progression may be determined by differential cellular susceptibility, related to mitochondrial function or proteostasis, differential cell-to-cell spread of pathology, or novel pathways and mechanisms. Risk factors determined from case-control studies indicate aetiological pathways and guide future preventive trials, but these may differ from risk factors that determine disease progression. Currently disease-modifying treatment trials focus on intervention in recently diagnosed patients, related to disease progression after diagnosis. Work on large-scale longitudinal cohorts over the last ten years has now enabled the collaborative study of large clinico-genetic datasets. Here we have carried out two progression GWASs: progression to mortality, and H&Y stage 3 or greater (H&Y3+). We have analysed data from 6766 PD patients with over 15,340 visits and mean follow-up ranging between 4.2 to 15.7 years.
Results
Overall 6766 participants with PD were analysed with mean follow-up between 4.2 and 15.7 years (Table 1). We did not have data from regular follow-up visits for all studies as some studies only contributed mortality data. However, in the studies that had regular follow-up visit data available, over 15,340 visits were analysed (Table 1).
GWAS of mortality
One study was excluded from the meta-analysis of mortality because the study-specific genomic inflation factor was above 1.2 and one study was excluded because less than 20 individuals reached the mortality endpoint (Supplementary Table 1).
5744 patients were included in the meta-analysis of mortality. Of these, 1846 (32.1%) individuals had died with a median time to death of 10.6 years from PD onset. The Kaplan-Meier curve for mortality stratified by cohort is shown in Supplementary Fig. 1. 7,696,389 Single Nucleotide Polymorphisms (SNPs) were present in at least 1,000 individuals and 7,313,918 SNPs passed meta-analysis filtering for heterogeneity and MAF variability. The genomic inflation value of the meta-analysis after filtering was 1.04.
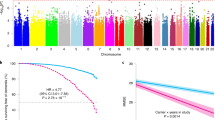
Two loci passed genome-wide significance and were identified to determine mortality in PD (Fig. 1). The top SNP was rs429358 in Chromosome 19 (p = 1.4 x 10−10), which tags the APOE ε4 allele (Table 2). This locus included 12 SNPs in total in linkage disequilibrium (r2 ≥ 0.6) with the independent significant SNP. One other locus in Chromosome 7 in TBXAS1 also reached significance (p = 7.7 x 10−10), and another locus in Chromosome 12 near SYT10 was nominally associated (p = 5.3 x 10−8). Regional association plots are shown in Fig. 1 and Supplementary Figs. 2–4. The top ten independent SNPs identified from GCTA-COJO and their nearest genes are reported in Table 2.

a The Manhattan plot showing two GWAS significant loci after meta-analysis. The blue dashed line indicates the threshold for genome-wide significance, p = 5 × 10−8. SNPs highlighted in red have p-value < 5 × 10−9. SNPs highlighted in orange have p-value < 5 x 10−8. One nominal association in Chromosome 12 is also annotated with the nearest gene, SYT10 (p = 5.3 × 10−8). b Forest plot for the top SNP rs429358 in Chromosome 19, in APOE. c Forest plot for the top SNP rs4726467 in Chromosome 7, in TBXAS1. d Forest plot for the top SNP rs10437796 in Chromosome 12, near SYT10. BP Base Pair, CI Confidence Interval, GWAS Genome Wide Association Study. HR Hazard Ratio. SNP Single Nucleotide Polymorphism.
We performed MAGMA gene- and gene-set analysis within FUMA (Functional Mapping and Annotation of Genome-Wide Association Studies; https://fuma.ctglab.nl/)7 to aggregate variant data to the level of whole genes or groups of genes8. In the MAGMA gene-based test, APOE was significantly associated with mortality (p = 1.9 x 10−10), and SYT10 was associated just below genome-wide significance (p = 3.6 x 10−6). No gene sets or tissues were significantly associated with mortality in the MAGMA analysis.
A locus in TBXAS1 was also significantly associated with PD mortality, with the top SNP rs4726467 and 5 additional SNPs in linkage disequilibrium (r2 ≥ 0.6). This SNP is an expression quantitative trait locus (eQTL), with the effect (minor) allele decreasing expression of TBXAS1 in the blood (eQTLGen; https://www.eqtlgen.org/)9 but not other tissues as reported in GTEx (https://gtexportal.org/). There was no evidence on GTEx that rs4726467 is a splicing Quantitative Trait Loci (sQTLs). Brain eQTL data at MetaBrain (https://www.metabrain.nl/)10 did not indicate this SNP was a significant cis-eQTL in any brain regions from European samples.
We also searched the public database LDproxy (https://ldlink.nci.nih.gov/) to look for coding variants that may be tagged by the top SNPs which could provide insight into the causal variants, genes, and their functions. We identified two coding variants in linkage disequilibrium (LD) within 500 kb of the top SNP in TBXAS1. One was a synonymous variant in HIPK2, rs34093649 (D’ = 0.24, R2 = 0.05, MAF 0.02) but this was not significant in the GWAS (p = 0.39). There was also one missense variant in PARP12, rs2286196, about 89 kb away from rs4726467 (D’ = 0.34, R2 = 0.01, MAF = 0.20), which was present in the GWAS meta-analysis with nominal significance (p = 0.03).
The top SNP in Chromosome 12, rs10437796, is not directly within SYT10 but increases SYT10 expression in the testis and decreases expression in the tibial nerve. The SNP also increases the expression of the long noncoding RNA (lncRNA) RP11-438D14.2 (ENSG00000259937) in the brain. This is a ‘sense intronic’ transcript to SYT10, a long non-coding transcript that is within an intron of a coding gene and does not overlap any exons. Brain cis-eQTL data from MetaBrain showed that the effect allele A of rs10437796 significantly increased the expression of SYT10 in the cortex but not in other brain regions. This SNP was not an eQTL or sQTL for any genes in the blood in eQTLGen. There were no coding variants in LD with this SNP in LDproxy within a 500 kb window.
We also performed colocalization analysis to determine whether the association signals for PD mortality and gene expression are driven by a shared causal variant (see Methods; Supplementary Table 2). We used cis-eQTL data from PsychENCODE and eQTLGen to examine gene expression in the whole brain or blood, respectively. However, no PD mortality loci showed evidence of colocalization with eQTLs (PP.H4 < 0.75; Supplementary Table 2).
We checked the top SNPs and genes (+/− 1 Mb) from the mortality GWAS in the most recent longevity GWAS11 to see if these were influencing PD-specific or more general population-based mortality and survival. APOE and specifically the ε4 tagging SNP, rs429358, was the strongest signal for longevity (beta = −0.05, p = 9.6 x 10−127). The TBXAS1 SNP, rs4726467, was not associated with longevity (p = 0.82). Other SNPs within or just outside the TBXAS1 gene boundaries were also not associated with longevity at genome-wide significance. The nearest associated SNP was rs149577943 which lay approximately 1 Mb outside of TBXAS1 (p = 6.1 x 10-5). The SYT10 SNP, rs10437796, was nominally associated with reduced longevity (beta = −0.008, p = 0.003) in the longevity GWAS.
There was evidence of sexual dimorphism for the APOE ε4 effect on PD mortality, similar to what was observed in the longevity study by Timmers et al. 11. We analysed the effect of the APOE ε4 SNP rs429358 in men and women separately in each cohort, then meta-analysed the results with a random-effects meta-analysis. Differences in the effect size in men vs. women were tested using the formula: (βmen – βwomen)/sqrt(SEmen2 + SEwomen2), where SE is the standard error of the effect estimate. This statistic follows the Z distribution. The effect of APOE ε4 on PD mortality was stronger in women than in men (betawomen = 0.54, SEwomen = 0.08 vs betamen = 0.23, SEwomen = 0.05, pdiff = 9.72 x 10-4).
GWAS of H&Y3+
3331 individuals across 5 cohorts were analysed for progression to H&Y3+ (Supplementary Fig. 5). 753 individuals (22.6%) met the outcome of H&Y3+, with a median time of 3.1 years. 6,549,622 SNPs passed filtering for heterogeneity and MAF variability. The genomic inflation factor of the meta-analysis was 1.03. The top ten independent SNPs from GCTA-COJO are reported in Table 3.
Four independent loci were significantly associated with progression to H&Y3+ (p < 5 x 10−8). Regional association plots for all GWAS significant loci are shown in Supplementary Figs. 6-9. The top locus was in Chromosome 1, with lead SNP rs115217673 within the MORN1 gene (Fig. 2) with 11 SNPs in total in the locus (r2 ≥ 0.6 with the independent significant SNP). The top SNP was not a significant eQTL for any genes, according to GTEx, eQTLGen, or MetaBrain databases.

Note that the loci in the Manhattan plot and the forest plots have been labelled with the physically closest genes, though these may not necessarily be the causal genes. a The Manhattan plot showing four GWAS significant loci after meta-analysis. The blue dashed line indicates the threshold for genome-wide significance, p = 5 × 10−8. SNPs highlighted in red have p-value < 5 × 10−9. SNPs highlighted in orange have p-value < 5 × 10−8. b Forest plot for the top SNP rs115217673 in Chromosome 1, in MORN1. c Forest plot for the top SNP rs145274312 in Chromosome 7, near ASNS. d Forest plot for the top SNP rs113120976 in Chromosome 4, near PDE5A. e Forest plot for the top SNP rs141421624 in Chromosome 2, in XPO1. BP Base Pair, CI Confidence Interval, GWAS Genome Wide Association Study. HR Hazard Ratio. SNP Single Nucleotide Polymorphism.
Published summary statistics from a previous progression GWAS by Iwaki et al.2 were downloaded from https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/ (accessed November 2019). Our top SNP in MORN1 was not associated with progression to H&Y3+ in the Iwaki summary statistics (beta = 0.61, p = 0.02). Meta-analysis with the Iwaki summary statistics after excluding overlapping cohorts did not reveal any genome-wide significant loci (see Supplementary Materials).
The second most significant locus was in Chromosome 7, with top SNP rs145274312, nearest to the ASNS gene. Only one SNP was included in this locus. This SNP was also not identified as an eQTL for any genes in the eQTL databases. It is important to note that this locus only included one variant with low MAF (1.1%), a wide confidence interval, and without supporting variants in LD (see Supplementary Fig. 9 and LDproxy (https://ldlink.nih.gov). This may be due to the LD patterns in the region as there are no variants in high LD (r2) with the lead variant, however, this locus should be interpreted with caution.
The third most significant locus was in Chromosome 4, with top SNP rs113120976. This was closest to the protein-coding gene PDE5A, however it is also near a long non-coding RNA LOC107986192 (NR_165235.1). The top SNP is a significant cis-eQTL in blood for USP53 with the effect allele T decreasing gene expression (eQTLGen). However, the SNP was not an eQTL in other tissues as reported in GTEx and MetaBrain. This locus included only two variants (Supplementary Fig. 8) and the lead variant had a low MAF of 1.3% so should also be interpreted with caution.
Finally, there was a locus in Chromosome 2 which was significantly associated with progression to H&Y3+, with top SNP rs141421624. This locus included 9 SNPs spanning several genes: XPO1, USP34, C2orf74, and KIAA1841, although only the top SNP reached GWAS significance. The lead SNP is intronic in the XPO1 gene. However, it is also a significant eQTL in blood for the genes KIAA1841 and AHSA2 (eQTLGen). It is also a significant eQTL in brain cortex for C2orf74, with the effect allele G increasing gene expression (MetaBrain).
In LDproxy (https://analysistools.cancer.gov/LDlink), there were also two missense variants within 500 kb in linkage disequilibrium with the lead SNP rs141421624. One of these is rs1729674, a missense variant in C2orf74, with a D’ of 1.0 R2 of 0.014, and MAF of 0.42. The other variant in LD with the lead SNP is rs76248080, a missense variant in the CCT4, D’ = 0.55. R2 = 0.014, MAF = 0.18. However, both of the missense variants were included in the GWAS meta-analysis and were not significantly associated with H&Y3+ (p > 0.05).
Colocalization analysis did not provide strong evidence for shared causal variants between PD H&Y3+ loci and gene expression, with no loci surpassing PP.H4 > 0.75 (Supplementary Table 3, Supplementary Fig. 10).
Candidate variant analysis
We did not find that any of the 90 PD risk SNPs were associated with PD progression at genome-wide significance (Supplementary Table 4). Two SNPs, representing rare LRRK2 (rs34637584) and GBA1 (rs114138760) variants, were not present in our analysis as we filtered out variants with MAF < 1%. Of the 88 PD risk variants tested, we found that only one variant, rs35749011, near KRTCAP2 and tagging the GBA1 p.E326K variant, was significantly associated with mortality (p = 3.6 x 10−4) passing the analysis-wide significance threshold (p-value threshold 0.05/88 = 0.00057). There was also one other variant, rs62333164 in CLCN3, which was nominally associated with progression to H&Y3+ (p = 0.005) (Supplementary Fig. 11).
We also examined the association between the PD GRS and progression in each cohort, adjusting for sex, age at onset, and PC1-PC5. The random-effects meta-analysis across cohorts did not show an effect of the GRS on mortality (HR = 0.99 [95% CI 0.95 to 1.04], p = 0.74), or H&Y3+ (HR = 1.02 [95% CI 0.96 to 1.08], p = 0.60).
We also analysed candidate variants that have been previously reported for progression (Supplementary Table 5). One previous study found that rs2242367, within the SLC2A13 gene and adjacent to the LRRK2 gene and PD risk locus, was associated with survival in Progressive Supranuclear Palsy (PSP)12. In our candidate variant analysis, this SNP rs2242367 was nominally associated with more rapid progression to mortality in PD in a meta-analysis across all cohorts (HR = 1.13 [95% CI 1.04 to 1.21], p = 0.002) (Supplementary Table 5). When we looked at association of this SNP in the QSBB cohort alone, where there is pathological confirmation of PD, there was no association between the PSP mortality SNP and PD mortality (HR = 1.13, p = 0.22).
Alzheimer’s Disease Genetic Risk Score (GRS)
To assess whether AD genetic risk outside of APOE influences mortality in PD, we created the AD GRS excluding the APOE region. In the random-effects meta-analysis, the AD GRS without APOE was only nominally associated with mortality (HR = 1.06 [95% CI 1.01 to 1.11], p = 0.03).
Power calculations
The power to detect a signal in a survival GWAS depends on a number of factors, including effect size, allele frequency of the effect allele, and the proportion of individuals meeting the outcome of interest. Using the ‘survSNP’ package13, we estimate that this study had 92% power to detect a significant effect (p < 5 × 10−8) for our top APOE SNP rs429358 in the mortality GWAS, given an allele frequency of 16%, Hazard Ratio of 1.34, event/death rate of 32.1% and median time to death of 10.6 years. Supplementary Fig. 12 shows how power changes with different event rates and allele frequencies. Clearly, power for progression studies will increase with longer follow-up as more individuals meet the outcomes.
For the top MORN1 SNP in the H&Y3 + GWAS, we had 87% power to detect a significant effect, given the allele frequency of 1.6%, Hazard Ratio of 2.76, event rate of 22.6%, and median time to event of 3.1 years. Supplementary Fig. 13 shows power at different event rates and allele frequencies.
Discussion
We have conducted a large meta-analysis GWAS of progression to clinical milestones in PD. We have identified loci in or near APOE, TBXAS1, MORN1, ASNS, PDE5A and XPO1 as relevant to survival and motor progression in PD. Using this single joint analysis instead of a two-stage replication approach has been shown to be more efficient and provides greater power14, and has been adopted by the most recent large-scale PD GWAS studies1,15,16,17. We found that the effects were largely consistent and replicated across cohorts, as illustrated in the forest plots and formal tests of heterogeneity.
The top hit for mortality was the APOE SNP rs429358 which tags the ε4 allele. APOE is the strongest genetic risk factor for AD18,19,20,21, and is also associated with cardiovascular disease22. In PD, APOE is associated with age at onset but this may be more generally related to aging, as the effect of APOE was similar in age of entry of controls23. Indeed, GWASs of longevity and survival in the general population have identified APOE as the strongest genetic factor, with the same ε4 (rs429358) allele associated with increased mortality24 and found less frequently in long-living individuals25. Thus, our finding that APOE is a risk factor for mortality in PD patients may not be specific to PD, as we have only examined all-cause mortality in this study. In addition, it can be difficult to precisely classify the cause of death26 as genetic variants could increase the vulnerability of PD patients to other causes of death, such as coronary heart disease. However, we also found that the APOE ε4 SNP rs429358 was nominally associated with progression to H&Y3+ (Supplementary Table 6). This could indicate that the APOE effect on mortality could be partly explained by motor progression and be PD-specific, since PD-related mortality could be due to falls and overall motor deterioration27,28.
We also analysed AD GRSs excluding APOE. There was no strong evidence that non-APOE AD genetic risk influences mortality in PD. We hypothesize that both dementia4 and mortality in PD are largely driven by amyloid-β pathology and plaque formation influenced by APOE ε4 genotype, although APOE may also contribute via other mechanisms such as immune responses18 and Lewy body pathology29,30.
APOE is also important for cognition and dementia in PD, and potentially motor progression31, but not PD risk1,32. In a separate GWAS study, we confirmed that APOE is a major risk factor for PD dementia4, and dementia in PD is predictive of later mortality33. Thus it is likely that APOE causes more rapid progression in PD as marked by both dementia and mortality, but we did not have the cause of death or cognition data in the majority of our cohorts to determine the extent to which PD dementia contributes to mortality.
The mortality GWAS also identified a locus in TBXAS1 which encodes a platelet aggregator and vasoconstrictor. This could be due to the influence of genetic factors for all-cause mortality, although these variants in TBXAS1 were not associated with survival/longevity in the general population11. We also identified a locus near SYT10 which regulates calcium-dependent exocytosis. Syt10 is also important for secretion of insulin-like growth factor-1 which has been suggested to play a role in the development of PD34 and deficits in dopamine neuron firing accompanied by motor problems35.
In our H&Y3 + GWAS, we identified 4 novel loci associated with progression, near MORN1, ASNS, XPO1, and PDE5A, although the variants near ASNS and PDE5A were only covered in two cohorts. In addition, the ASNS and PDE5A loci included only single variants, and the variants had low MAF (~1%) and wide confidence intervals so should be interpreted with caution. There is a risk that these may be false positives and replication in other cohorts is needed. We have made our summary statistics available at https://tinyurl.com/PDprogressionv2 to encourage replication efforts and enable meta-analysis. Gene ontology analysis did not reveal any biological pathways, gene sets, or tissues that were enriched among the GWAS hits, and this is likely because we are still underpowered for this analysis. A more in-depth discussion of candidate genes is provided in the Supplementary Materials.
There did not appear to be overlap in the hits identified in the mortality and H&Y3+ GWASs (Supplementary Materials). This may be because there are differences in the genetic variation contributing to different forms of progression. For example, motor and cognitive progression in PD are moderately but not perfectly correlated (r = 0.35)3 and different PD subtypes show different rates of motor and cognitive impairment/progression36,37. Thus is it possible that the different findings from the two GWASs may reflect a divergence in the genetic underpinnings for different types of progression. Alternatively, it could reflect the incomplete sample overlap and different sample sizes between the two GWASs.
In line with previous PD progression GWASs, the majority of the 90 PD risk SNPs were not associated with PD progression2,3,5,38. Although LRRK2 G2019S has been previously associated with slower progression39, this variant did not meet our allele frequency filter. Overall, the lack of association of risk variants with progression is an important finding in itself which has also been replicated in other PD progression GWASs2,3,5,38, and suggests that other factors and pathways may influence progression after disease onset, e.g. amyloid pathology appearing at later disease stages.
We showed that one variant, rs35749011, linked to GBA1 p.E326K (also known as p.E365K) was associated with mortality. Interestingly this variant does not cause Gaucher disease or have a major effect on glucosylceramide levels suggesting a dissociation between glucosylceramide and the role of GBA1 in PD progression. This is consistent with the recently reported trial data reporting a lack of effect of the glucosylceramide synthase inhibitor venglustat in modifying PD progression40, although we only examined common GBA1 variants in this study and rarer variants may have a larger effect.
We were not able to replicate findings for other candidate variants nominated from previous PD progression GWASs. We examined results for rs382940 in SLC44A1 for progression to H&Y3 + 2, however, this was not associated with progression in any of our results.
We also did not identify a mortality or motor progression effect for variants in RIMS2, WWOX, and TMEM108 which have been reported for PD dementia5. The p-values for these variants were all > 0.3 in our GWASs (Supplementary Table 5) although we did not analyse cognitive impairment or dementia in this study. Overall, our study has identified novel associations and largely not replicated previous GWAS progression findings apart from APOE2,3,4,5. Part of this may be due to the different measures of PD progression (e.g. early and late-stage motor progression, cognitive progression, mortality, and dementia). It is likely that different forms of PD progression are influenced by different genetic variants and processes, although there may be some shared factors such as APOE. Furthermore, the heterogeneity of the cohorts (e.g. disease stage, inclusion criteria), in addition to the difficulty in measuring progression through clinical scales, may explain why we have not replicated previous findings even for the same outcome measures as previous studies, namely H&Y3+. We have shown replication of effects across our cohorts, however, replication in independent cohorts of all genetic loci reported in our study and previous studies is necessary.
We did not find evidence to support APOE ε2 and MAPT H1 haplotype as factors for mortality. We found some evidence suggesting the PSP mortality SNP, rs224236712, was also associated with more rapid mortality in PD. This finding could indicate that there is some misclassification of PSP cases in our PD cohorts, as PSP can be frequently misdiagnosed as PD and we did not have pathological diagnosis data on the majority of cases. We found no association between this SNP rs2242367 and PD mortality in the QSBB cohort, where there is pathological confirmation of PD – thus we cannot rule out the possibility that there is ‘contamination’ of PSP cases in the other cohorts which do not have pathological diagnoses available. Alternatively, the regulatory effects of this SNP, which is separate from the LRRK2 PD risk locus, may influence survival in both PD and PSP.
This study is one of the largest GWASs of PD progression and the first large-scale GWAS of PD mortality. However, larger sample sizes and longer follow-ups are needed to detect variants with smaller effects (e.g. HR < 1.2) or lower allele frequencies. Due to our limited sample size, we are still underpowered to detect some loci influencing disease progression, particularly in the H&Y3 + GWAS which had a smaller sample size. Further replication efforts are needed in independent cohorts from other parts of Europe and North America. In addition, given the low MAF of some of the top variants identified particularly in the H&Y3 + GWAS, rare variant and burden analysis in sequencing data would be useful to evaluate the impact of rare variants in candidate genes. This was not feasible in the current study as the majority of cohorts only had SNP array data.
We also removed SNPs with heterogeneous effects across cohorts, following previous studies2, and to ensure reproducible and robust results, with the aim to remove SNPs that may be false positive results. It is possible that some true progression SNPs have been excluded with this conservative approach.
A second key limitation is that our findings are derived only from individuals of European ancestry and may not extend to individuals from other populations. It has been shown that PD genetic risk factors can differ or have heterogeneous effects in different ancestries15,16,17, and this may also be the case with PD progression variants.
Thirdly, more data is needed on post-mortem pathological diagnosis to conduct cause-of-death analyses, as some of the mortality variants may relate to general population mortality rather than having a specific effect on PD progression. However, APOE is the only gene identified in our mortality GWAS that overlaps with general population longevity GWAS. Another limitation is the heterogeneity between cohorts and PD case selection. Our cohorts tend to be recruited from specialist clinics and groups of patients, and this may lead to a tendency to recruit more atypical patients, or rapidly progressing patients. More population-based studies are needed to improve generalisability of these results. Several of our cohorts are also non-incident, with a delay between symptom onset and study entry, and this means that we are not able to capture the most rapidly progressing patients.
In addition, we nominated genes from the top SNPs based on physical proximity and eQTL databases, however, additional fine-mapping and annotation are needed to prioritise causal variants and genes for each locus. More in-depth polygenic risk score analysis, including SNPs below genome-wide significance, could also be performed to examine the overlap between PD risk, progression, AD, and other phenotypes however we were not sufficiently powered to conduct in-depth permutation testing of p-value thresholds in this study. Finally, the interpretation of GWAS for neurological disease remains limited by the resolution of the effects of genomic variants on gene expression from bulk RNA sequencing studies. Rapidly increasing sample sizes, and the development of single-cell resources will enable a more direct interpretation of the relationship between genomic variants and disease biology.
One major challenge is that clinically, motor progression is heterogeneous and influenced by multiple factors, such as the presence and severity of non-motor symptoms, genetic variation, medication, and comorbidities such as Type 2 Diabetes. The aim of the current study was to identify genetic determinants and understand more about the biology of PD progression, thus we did not adjust for other factors that may influence progression as this may be overcorrection. It is possible that some of these factors may lie on the causal pathway between genotype and clinical progression as intermediary factors41. We also did not have available data in the current cohorts to include these in models. However future studies which aim to predict PD progression could adjust for risk and protective factors for more accurate prediction.
We conducted two large-scale GWASs of PD progression, including the first GWAS of mortality in PD. We identified six significant genome-wide signals, including TBXAS1. We also showed that the genetic factors influencing progression in PD are largely different to those influencing PD risk, emphasising the need for further studies of progression. This work will help us to better understand the biology of PD progression and develop new disease-modifying treatments.
Methods
Brief methods
We studied 11 cohorts from Europe and America, and included cohorts in each analysis who had sufficient data on the outcomes of interest (see below for further details). Genotyping, quality control, and imputation was performed in each cohort separately but following the same steps. Only variants with high imputation quality scores (INFO/R2 > 0.8) and minor allele frequency (MAF) > 1% were retained for analysis.
We assessed the following two clinical outcomes: mortality, and H&Y3+. H&Y stage 3 is an important clinical milestone and a commonly used outcome to measure motor progression, as it marks the onset of postural instability42, usually accompanied by falls. It is associated with a more rapid stage of motor progression and more severe disability43,44,45. Cohorts were excluded if less than 20 individuals met the outcome of interest during the follow-up period, or < 5% of the total cohort size. For the mortality GWAS, all the cohorts were analysed with the exception of the Parkinson’s Progression Markers Initiative (PPMI): Tracking Parkinson’s, Oxford Discovery, Queen Square Brain Bank (QSBB), CamPaIGN, Cambridge PD cohort, UK Biobank incident cases, UK Biobank prevalent cases, Drug Interaction With Genes in Parkinson’s disease (DIGPD), Trondheim, and Oslo. The PPMI cohort was excluded from the mortality GWAS as less than 20 deaths were recorded in the data download (14/08/2019). For the GWAS of H&Y3+, we analysed cohorts which had this data available: Tracking Parkinson’s, Oxford Discovery, PPMI, DIGPD, and Oslo.
Progression to the clinical milestones was analysed using Cox proportional hazard models. Progression to mortality was assessed from the starting timepoint of PD motor symptom onset, except in the UK Biobank cohorts where PD diagnosis was used (see below). Progression to H&Y3+ was assessed from the time of study entry (baseline visit). We used study entry for progression to H&Y3+ as this is the period where participants can be observed and measured, whereas this would not be possible if disease onset was used, particularly since several of our cohorts were not incident cohorts. Individuals who met the outcome of H&Y3+ at study entry were left censored and excluded from analyses. In each model, we adjusted for age at onset, sex, and the first five genetic principal components to adjust for population stratification. Meta-analysis was performed in METAL (version 2011-03-25)46, using an inverse variance weighted fixed effects model. GWASs with a genomic inflation factor above 1.2 were excluded from the meta-analysis. Only SNPs that were genotyped/imputed in > 1000 individuals across all cohorts were included in the final results. SNPs with heterogeneous effects across cohorts were also excluded (p-value < 0.05 for Cochran’s Q-test for heterogeneity, and/or I2 > 80). The null hypothesis was tested with the standard GWAS significance level of 5 x 10-8. Results were uploaded to Functional Mapping and Annotation of GWAS (FUMA; https://fuma.ctglab.nl/)7 to annotate, prioritise, and visualize GWAS results, and perform gene-set analysis with MAGMA. Genome-wide Complex Trait Analysis conditional and joint analysis (GCTA-COJO, version 1.94.1) was used to identify independently-associated SNPs47, using all individuals in the Accelerating Medicines Partnership Parkinson’s disease (AMP-PD) whole genome sequencing dataset as a reference sample (N = 9422, including PPMI). Fine-mapping of the top loci was performed with Probabilistic Annotation INtegraTOR (PAINTOR) (https://bogdan.dgsom.ucla.edu/pages/paintor/) following the recommended pipeline, to prioritize causal variants and integrate functional genomic data48,49,50. Forest plots for the top SNPs were generated in R v3.6 using the forestplot package, enabling us to determine consistency, and replication of survival risk alleles across cohorts. Further details are provided below.
We also performed candidate variant analysis of the 90 PD risk variants from the most recent PD case-control GWAS1, and the cumulative PD genetic risk score (GRS). The GRS was created using only the 90 independent genome-wide significant variants from Nalls et al. 1, without any further clumping and thresholding. We also examined associations for other candidate variants that have been reported in PD and Progressive Supranuclear Palsy (PSP) progression: SLC44A12, RIMS2, WWOX, TMEM1085, APOE ε2 allele, MAPT H1 haplotype, and rs2242367 adjacent to the LRRK2 locus12. We analysed the Alzheimer’s disease (AD) GRS in relation to PD progression. 38 loci passing genome-wide significance from the latest AD GWAS were used to create the AD GRS19. The APOE region was excluded from the GRS (hg19/GRCh37 coordinates 19:40,000,000-50,000,000)19.
Cohorts
We studied 12 cohorts from Europe and America: Tracking Parkinson’s51 (ClinicalTrials.gov identifier NCT02881099), Oxford Discovery52, Parkinson’s Progression Markers Initiative (PPMI; NCT01141023)53, Queen Square Brain Bank (QSBB) pathologically-confirmed PD cases, Calypso41,54, UK Biobank incident cases and UK Biobank prevalent cases (see below for details), Cambridgeshire Parkinson’s Incidence from GP to Neurologist (CamPaIGN)55,56, the Cambridge PD Research Clinic cohort32,57, Drug Interaction With Genes in Parkinson’s Disease (DIGPD; NCT01564992)58, the Trondheim Parkinson’s Disease study (Trondheim)59, and the Oslo Parkinson’s Disease study (Oslo)60. All participants provided written informed consent. A brief summary of key cohort characteristics and inclusion/exclusion criteria are provided below.
Cohorts were excluded if less than 20 individuals met the outcome of interest during the follow-up period, or < 5% of the total cohort size. Small numbers can produce unreliable effect size estimates and extremely wide confidence intervals. All cohorts were included for analysis of mortality, with the exception of the PPMI study as not enough patients met the outcome. For the analysis of other clinical outcomes, not all cohorts had available clinical assessments. For Hoehn and Yahr stage, we analysed data from Tracking Parkinson’s, Oxford Discovery, PPMI, DIGPD, and Oslo.
If data was available within a cohort, participants who were known to be re-diagnosed with a non-PD condition were excluded from analyses.
Tracking Parkinson’s
Tracking Parkinson’s is a longitudinal, prospective, multi-centre observational study in the UK51. Patients were recruited at 72 secondary healthcare centres that are part of the UK National Health Service. Participants were required to have a clinical diagnosis of PD according to Queen Square Brain Bank criteria and supported by neuroimaging if the clinical diagnosis was uncertain. PD participants were also required to be aged between 18 and 90 years, and be diagnosed with PD within 3.5 years of recruitment. Both drug-naive and treated patients were eligible. Patients were assessed at 6-month intervals with a standardised battery of clinical assessments every 18 months. Participants were excluded if they had severe comorbid illness that interfered with participation in clinical visits, other degenerative forms of parkinsonism (e.g. progressive supranuclear parkinsonism), or parkinsonism due to significant cerebrovascular disease.
Oxford Discovery
Oxford Discovery is a prospective, longitudinal study led by the Oxford Parkinson Disease Centre52. Participants with early idiopathic PD were recruited from neurology clinics across the Thames Valley area in the UK. Participants were required to be diagnosed with PD within the last 3 years according to the UK PD Brain Bank criteria by a neurologist or geriatrician with a PD special interest. Participants were excluded if they had non-idiopathic parkinsonism, dementia before PD by 1 year suggesting Dementia with Lewy Bodies, or cognitive impairment that prevented them from providing informed consent. Participants are assessed every 18 months.
PPMI
The Parkinson’s Progression Markers Initiative (PPMI) study is a longitudinal, observational study of early PD patients at multiple sites across the US and Europe (https://www.ppmi-info.org/)53. PD participants were required to have two of the following symptoms: resting tremor, bradykinesia, and rigidity (must have either resting tremor or bradykinesia), or either asymmetric resting tremor or asymmetric bradykinesia. Participants were required to be diagnosed with PD less than 2 years before screening, aged 30 years or older at the time of PD diagnosis, Hoehn and Yahr stage I or II at baseline, confirmation of dopamine transporter deficit from dopamine transporter single photon emission computed tomography (DAT-SPECT) scan and not expected to require PD medication within at least 6 months from baseline. Participants were excluded if they were already taking a PD medication or had taken taken levodopa, dopamine agonists, MAO-B inhibitors or amantadine within 60 days of Baseline, or had ever taken levodopa or dopamine agonists before baseline for more than 60 days in total. Participants were assessed with the full battery of assessments every 12 months. PPMI data was downloaded on 14/08/2019.
Queen Square Brain Bank
The Queen Square Brain Bank for Neurological Disorders (QSBB) is an archive of brains and tissue from individuals with neurodegenerative disease and neurologically normal controls. The QSBB is based at University College London in London. A request for clinical data and DNA from pathologically-confirmed PD patients was submitted in May 2018.
Calypso
Calypso is a community-based prevalence study of PD in Cardiff, Wales. Patients were referred by neurologists, geriatricians, and specialty PD nurses, as well as by self referral54. Participants were either invited to a research clinic or could remotely complete questionnaires. Participants who attended the clinic were assessed according to the Queen Square Brain Bank diagnostic criteria for PD, whereas remote participants had their clinical notes reviewed to confirm their diagnosis.
UK Biobank
PD cases were identified from UK Biobank from hospital episode statistics (HES) with an ICD10 code (G20 for PD) in either the primary or secondary position. PD patients were also identified from self-report and death records. UK Biobank data was downloaded on 13/06/2020 (application 46450). PD patients were classified as either prevalent, incident, or undefined, following the ‘Definitions of Parkinson’s Disease and the major causes of Parkinsonism: UK Biobank Phase 1 Outcomes Adjudication’ document (version 1.0, March 2018; http://biobank.ctsu.ox.ac.uk/showcase/showcase/docs/alg_outcome_pdp.pdf). Briefly, prevalent cases were defined as PD patients who had the first PD ICD code date prior to the baseline assessment, or self-reported PD at the baseline assessment. Incident cases were defined as patients with PD detected by HES with the PD ICD code date after the date of baseline assessment. Patients with PD coded in any position in the death register records, but did not have PD in the HES records at any point were also defined as incident cases but these patients were excluded from our analysis. Patients who did not self-report PD at baseline but at a later follow-up visit, and who did not have PD in any HES records or death register records were classified as ‘undefined’. These patients were also excluded from analysis. In our study, we analysed prevalent and incident PD cases separately.
The date of PD diagnosis was defined according to UK Biobank guidelines, using the earliest date of the PD code from HES or self-report. This date of diagnosis was used as a proxy for PD onset in analysis.
Version 2 of the UK Biobank genotype data was used. Quality control and imputation as described below was performed only in the subset of PD cases in the UK Biobank, rather than existing data on the whole cohort.
CamPaIGN
The Cambridgeshire Parkinson’s Incidence from GP to Neurologist (CamPaIGN) study is an observational, longitudinal study of incident PD patients in the county of Cambridgeshire, UK55,56. The study is an unbiased and population-representative incident PD cohort. New cases of parkinsonism between 2000 and 2002 in Cambridgeshire were referred to the study through multiple sources (general practitioners, neurologists, geriatricians, PD specialist nurses, and hospital discharge coding departments). Cases with suspected onset of parkinsonism prior to the study were excluded. For PD participants, the UK Parkinson’s Disease Brain Bank criteria were used to confirm the diagnosis. Patients were followed up annually and assessed with a standardized battery of demographic, disease history, and neurological assessments.
Cambridge PD research clinic
PD patients were recruited from the PD Research Clinic at the Cambridge Centre for Brain Repair32,57. Participants were required to meet the UK Parkinson’s Disease Society Brain Bank diagnostic criteria. Participants completed a comprehensive battery of clinical and neuropsychological tests, the same as used in the CamPaIGN study, on at least one occasion.
DIGPD
The Drug Interaction With Genes in Parkinson’s disease (DIGPD) study is a longitudinal cohort study in France58. Patients with PD meeting the UK Parkinson’s Disease Society Brain Bank criteria were consecutively recruited from 2009 to 2013 in 4 French university hospitals and 4 general hospitals. PD participants were required to have a disease duration of 5 years or less at recruitment. Patients were followed up longitudinally and assessed with standardised clinical assessments and questionnaires every year for 5 years.
Trondheim
The Trondheim PD cohort is a prospective longitudinal study of PD patients at the Department of Neurology at St. Olav’s Hospital in Trondheim, Norway59. Patients were followed from 1997 onwards, with some having been followed since 1980. Sequential new referrals, over the age of 22 years, were referred to the study. The majority of the participants (80%) resided within 50 miles of Trondheim. PD participants were required to have at least two of the three cardinal signs (resting tremor, bradykinesia, and rigidity), improvement through adequate dopaminergic therapy, and the absence of atypical features or other causes of parkinsonism. Participants with atypical disease presentation were excluded, mainly after autopsy. Other than this, there were no other exclusion criteria other than age. Death records were linked to the Norwegian Cause of Death registry and the Cancer Registry of Norway.
Oslo
The Oslo PD cohort is a cohort of patients recruited from a movement disorders unit at Oslo University Hospital60. Participants were required to have a clinical diagnosis of PD by a neurologist. As a large proportion of referrals to the movement disorders unit are for evaluation for advanced treatment options, such as Deep Brain Stimulation, the patients in this study tend to have an earlier age at onset, severe motor fluctuations, good levodopa response and better cognitive function perhaps than other PD cohorts60. Clinical data was collected by assessing patients in the outpatient clinic or from retrospective medical records. Participants were assessed when possible during their regular outpatient clinic appointments, so the mean time between assessments in this dataset was 1.7 years. Death records were obtained from the Norwegian National Registry.
Genotyping quality control and imputation
Genotyping, quality control, and imputation were performed in each cohort separately but following the same steps. Standard quality control procedures were performed in PLINK v1.9 to remove low-quality variants, samples, related individuals, and ancestry outliers. Briefly, individuals with low overall genotyping rates (<98%), related individuals (Identity-By-Descent PIHAT > 0.1), and heterozygosity outliers (>2 standard deviations away from the mean) were removed. Individuals whose clinically reported biological sex did not match the genetically determined sex were also removed.
To remove ancestry outliers, Principal Components Analysis (PCA) was conducted on a linkage-disequilibrium (LD) pruned set of variants (removing SNPs with an r2 > 0.05 in a 50 kb sliding window shifting 5 SNPs at a time) after merging with European (CEU) samples from the HapMap 3 reference panel. Individuals who were more than 6 standard deviations away from the mean of any of the first 10 principal components were removed.
Variants were removed if they had a low genotyping rate (< 99%), Hardy-Weinberg Equilibrium p-value < 1 x 10-5, or minor allele frequency < 1%.
Following quality control, genotypes from each cohort were imputed separately using the Michigan Imputation Server. All cohorts were imputed to the Haplotype Reference Consortium panel (r1.1). Only variants with high imputation quality scores (INFO/R2 > 0.8) were retained for analysis, and imputation dosages were converted into hard call genotypes.
Related and duplicated individuals across cohorts were identified by merging individual-level genotype data. One individual from each pair of related individuals was removed (PIHAT > 0.1).
Statistical analysis
We assessed the following clinical outcomes: mortality, and Hoehn and Yahr stage 3 or greater (when postural instability is present).
The time to event was taken as the first visit where the outcome was met. Individuals who were missing data at all timepoints for the assessment were excluded (e.g. if Hoehn and Yahr stage data was missing at all visits, that patient was excluded from the analysis of progression to Hoehn and Yahr stage 3+).
Progression to each clinical milestone from PD onset was assessed using Cox proportional hazard models, adjusting for age at onset, gender, and the first 5 genetic principal components to adjust for population stratification. For mortality, PD onset was used as the starting timepoint. For Hoehn and Yahr stage 3 or greater, the starting timepoint was set as study entry/baseline visit. We report the proportional hazards assumption p-values for each of the top 10 SNPs in each cohort in Supplementary Tables 7 and 8. Analysis was performed in R using the survival package.
Meta-analysis and visualization
Meta-analysis was performed in METAL, using an inverse variance weighted fixed effects model. GWASs with a genomic inflation factor above 1.2 were excluded from the meta-analysis. Genomic control correction was used to adjust the overall alpha error. After meta-analysis, only SNPs that were present in > 1000 individuals were included in the final results. SNPs with heterogeneous effects across cohorts were also excluded (p-value < 0.05 for Cochran’s Q-test for heterogeneity, and/or I squared > 80). Variants with MAF variability greater than 15% across the cohorts were also excluded. The null hypothesis was tested with the standard GWAS significance level of 5 x 10-8.
Results were uploaded to Functional Mapping and Annotation of GWAS (FUMA; https://fuma.ctglab.nl/)7 to annotate, prioritise, and visualize GWAS results. Standard settings were used in FUMA, with the exception of a higher maximum p-value of to identify lead SNPs (5 x 10-5) so that we could report nominal associations. eQTL gene mapping using all tissue types was also used, in addition to positional mapping. Gene and gene-set analysis was performed with MAGMA within FUMA. Forest plots were generated in R v3.6 using the forestplot package.
GCTA-COJO
Genome-wide Complex Trait Analysis conditional and joint analysis (GCTA-COJO version 1.94.1,https://yanglab.westlake.edu.cn/software/gcta/#COJO) was used to identify if there were multiple independent SNPs within the same locus47,61. It performs a stepwise model selection procedure to select independently associated SNPs47,61.
GCTA-COJO requires a reference sample to estimate LD correlations between SNPs. For our reference sample, we used whole genome sequencing data from the Accelerating Medicines Partnership Parkinson’s disease (AMP-PD), including both PD cases and healthy controls. Standard quality control filters were applied to the AMP-PD data, as described previously4 and outlined here. Samples were removed if they had a call rate < 98%, excess heterozygosity (> 2 standard deviations from the mean heterozygosity rate), mismatching clinical sex and genetically determined sex from X chromosome heterogeneity, or if they were from related individuals (Identity-By-Descent PIHAT > 0.125). Variants were excluded if they had missingness > 5%, minor allele frequency < 1%, or Hardy-Weinberg Equilibrium p-value < 1 x 10−5. To remove ancestry outliers, PCA was conducted on a LD-pruned set of variants after merging with CEU + TSI samples from the HapMap 3 reference panel. Individuals who were more than 6 standard deviations away from the mean of any of the first 10 principal components were removed. After all quality control filters had been applied, 9422 individuals were remaining in the AMP-PD dataset.
AMP-PD data was in genome build hg38 and lifted over to hg19/GRCh37 genome build using liftOver (RRID:SCR_018160; https://genome.sph.umich.edu/wiki/LiftOver) to match the build of the GWAS summary statistics.
Colocalization
We performed colocalization analyses using Coloc version 5.1 (https://chr1swallace.github.io/coloc/index.html)62. We also used the package colochelpR to help prepare datasets for use in coloc (https://github.com/RHReynolds/colochelpR, https://doi.org/10.5281/zenodo.5011869)63. We used cis-eQTLs in blood from eQTLGen (https://www.eqtlgen.org/cis-eqtls.html)9, and cis-eQTLs in the brain from PsychENCODE (http://resource.psychencode.org/)64. Both datasets were downloaded on 22/03/2022.
We followed the same method as in Krohn et al. 65,66. For colocalization analysis (https://github.com/RHReynolds/RBD-GWAS-analysis/). For each locus, we examined all genes with 1 Mb of a significant locus in the PD mortality GWAS (p < 5 x 10−8). Coloc was run using default priors. These are the prior probabilities that any random SNP in the region is associated with trait 1 or trait 2, p1 = 10-4 and p2 = 10-4. We used a threshold of p12 = 5 x 10−6 for the p12 prior, which is the probability that a SNP in the region is associated with both traits. Loci with a posterior probability of hypothesis 4 (PP.H4) ≥ 0.75 were considered colocalized due to a single shared causal variant, rather than two distinct causal variants (PP.H3).
Fine-mapping with Probabilistic Annotation INtegraTOR (PAINTOR)
The top 10 independent variants from each GWAS (Table 2 and Table 3) were selected for statistical fine-mapping with PAINTOR v3.048,49,50. We followed the recommended pipeline at the PAINTOR v3.0 wiki (https://github.com/gkichaev/PAINTOR_V3.0/wiki) and which has been used in other GWASs67. Firstly, a region of 50 kb around the most significant SNP was selected (+/− 25 kb). Z-scores were calculated from the GWAS summary statistics beta effect size and p-value:
where Φ−1 is the inverse cumulative distribution function of the normal distribution. Z-scores were calculated in R using the zsc function from the dotgen package. Secondly, linkage disequilibrium was computed from the 1000 Genomes (Phase 3) reference data for each of the loci. Thirdly, an annotation matrix was created for each locus using all the annotations in the annotation library provided by PAINTOR. Finally, PAINTOR was run on all loci together, on each annotation independently. This was done using the default settings in PAINTOR, which performs approximate inference and enumeration under the assumption of 2 causal variants per locus. The fine-mapped variants with posterior probability > 0.9 are reported inSupplementary Tables 9 and 10. However, it is important to consider that statistical fine-mapping methods are limited and may not necessarily identify causal SNPs in all GWAS loci; functional validation is required to confirm candidate variants.
PD risk SNPs and GRS
We also performed candidate variant analysis of the 90 PD risk SNPs from case-control GWAS1, and the PD genetic risk score (GRS). The GRS is a cumulative risk score for each individual created from the sum of the genome-wide significant PD risk alleles weighted by effect size. The GRS was created in PLINK v1.9 using the 90 independent genome-wide significant variants from Nalls et al. 1. The standardised risk score was analysed in each cohort for each outcome using Cox proportional hazard models, adjusting for age at onset, sex, and the PC1-PC5. We created and tested the GRS in each cohort separately and then meta-analysed results using random-effects meta-analysis in R using the package meta.
Candidate variant analysis
We also examined associations for other candidate variants that have been implicated in PD progression. Previous large-scale genome-wide association studies have identified variants in SLC44A1 for progression to Hoehn and Yahr stage 3 or greater2, and variants in RIMS2, WWOX, and TMEM108 for progression to dementia5. We also examined results for rs7412 tagging the APOE ε2 allele, rs8070723 tagging the MAPT H1 haplotype, and rs2242367 adjacent to the LRRK2 locus, which was associated with survival in Progressive Supranuclear Palsy (PSP)12. We extracted the results for these 7 SNPs from each of our progression GWAS meta-analysis results. We applied Bonferroni correction to adjust for multiple testing for the number of variants tested, with p-value threshold p = 0.05/7 = 0.007.
Longevity GWASs
To help clarify whether our mortality GWAS results were specific to PD mortality or more general mortality/survival, we searched the most recent longevity GWAS11. and the GWAS Catalog (https://www.ebi.ac.uk/gwas/). Summary statistics from the Timmers et al. 11. Longevity GWAS were downloaded from https://datashare.ed.ac.uk/handle/10283/3599. We searched these summary statistics for the top SNPs and genes (+/− 1 Mb) from our PD mortality GWAS results.
Ethics
Tracking Parkinson’s: The West of Scotland Research Ethics Service (WoSRES) Research Ethics Committee gave ethical approval for this study (ref 11/AL/0163). Oxford Discovery: NRES Committee, South Central Oxford A Research Ethics Committee gave ethical approval for this study (ref 16/SC/0108). CamPaIGN: Cambridge Research Ethics Committee gave ethical approval for this study. Cambridge PD Research Clinic: Cambridge Research Ethics Committee gave ethical approval for this study. PPMI: The Research Subjects Review Board at the University of Rochester approved the PPMI study protocol. UK Biobank: UK Biobank has approval from the North West Multi-centre Research Ethics Committee (MREC) as a Research Tissue Bank (RTB). QSBB: The London Central Research Ethics Committee gave ethical approval for this research tissue bank (ref 18/LO/0721). DIGPD: The Ethical Committee Ile-De-France VI gave ethical approval for this study (ID project: 2009-A00109-48). Calypso: Wales Research Ethics Committee 3 gave ethical approval for this study. Trondheim: the Ethics Committee of Central Norway gave ethical approval for this study (ref 2011/1137). Oslo: the Regional Committee for Medical Research Ethics in South-East Norway gave ethical approval for this study.
Data availability
GWAS summary statistics are available for download at: https://tinyurl.com/PDprogressionv2 (https://doi.org/10.5281/zenodo.8017385). Tracking Parkinson’s data is available through the Tracking Parkinson’s portal: https://www.trackingparkinsons.org.uk/about-1/data/. The Oxford Parkinson’s Disease Centre Discovery Cohort data (https://doi.org/10.1016/j.parkreldis.2013.09.025) are available on request (Michele Hu, [email protected]). The Cambridgeshire Parkinson’s Incidence from GP to Neurologist (CamPaIGN) (https://www.thebarkerwilliamsgraylab.co.uk/parkinsons-disease/current-studies-pd/) data and the Cambridge clinic data are available on request (Caroline Williams-Gray/ Roger Barker; [email protected]). Parkinson’s Progression Markers Initiative (PPMI) data was accessed from the PPMI platform: https://www.ppmi-info.org/access-data-specimens/download-data. Queen Square Brain Bank for Neurological Disorders (QSBB) data are available upon request ([email protected]). UK Biobank data were accessed through the UK Biobank: https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access. Drug Interaction With Genes in Parkinson’s Disease (DIGPD) data (https://clinicaltrials.gov/ct2/show/NCT01564992) are available upon request (Jean-Christophe Corvol, [email protected]). Calypso data are available on request to the study team (Huw Morris, [email protected]). The Trondheim Parkinson’s Disease Study (Trondheim) data are available on request (https://doi.org/10.14802/jmd.21029). The Oslo Parkinson’s Disease data are available on request (Lasse Pihlstrom/ Mathias Toft, [email protected]).
Code availability
All code for our analyses is publicly available at https://github.com/manuelatan/PD-survival-GWAS (https://doi.org/10.5281/zenodo.7923843).
References
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18, 1091–1102 (2019).
Iwaki, H. et al. Genomewide association study of Parkinson’s disease clinical biomarkers in 12 longitudinal patients’ cohorts. Mov. Disord. 1–12 https://doi.org/10.1002/mds.27845 (2019).
Tan, M. M. X. et al. Genome-wide association studies of cognitive and motor progression in Parkinson’s disease. Mov. Disord. https://doi.org/10.1002/mds.28342 (2020).
Real, R. et al. Association between the LRP1B and APOE loci and the development of Parkinson’s disease dementia. Brain 146, 1873–1887 (2023).
Liu, G. et al. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson ’ s disease. Nat. Genet. 53, 787–793 (2021).
Fagan, E. S. & Pihlstrøm, L. Genetic risk factors for cognitive decline in Parkinson’s disease: a review of the literature. Eur. J. Neurol. 24, 561–e20 (2017).
Watanabe, K., Taskesen, E., Van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1–10 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, 1–19 (2015).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Klein, et al. Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain-related diseases. bioRxiv 2021.03.01.433439 (2021).
Timmers, P. R. H. J., Wilson, J. F., Joshi, P. K. & Deelen, J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. Nat. Commun. 11, 1–10 (2020).
Jabbari, E. et al. Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol 20, 107–116 (2021).
Owzar, K., Li, Z., Cox, N. & Jung, S. H. Power and sample size calculations for SNP association studies with censored time-to-event outcomes. Genet. Epidemiol. 36, 538–548 (2012).
Skol, A. D., Scott, L. J., Abecasis, G. R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 209–213 (2006).
Kim, J. J. et al. Multi-ancestry genome-wide meta-analysis in Parkinson’s disease. medRxiv https://doi.org/10.1101/2022.08.04.22278432 (2022).
Rizig, M. et al. Articles Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: a genome-wide association study. Lancet Neurol. https://doi.org/10.1016/S1474-4422(23)00283-1 (2023).
Foo, J. N. et al. Identification of risk Loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: A genome-wide association study. JAMA Neurol 77, 746–754 (2020).
Mahoney-Sanchez, L., Belaidi, A. A., Bush, A. I. & Ayton, S. The complex role of Apolipoprotein E in Alzheimer’s disease: an overview and update. J. Mol. Neurosci. 60, 325–335 (2016).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Mahley, R. W. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 94, 739–746 (2016).
Blauwendraat, C. et al. Parkinson’s disease age at onset genome‐wide association study: Defining heritability, genetic loci, and α‐synuclein mechanisms. Mov. Disord. 34, 866–875 (2019).
Rosvall, L. et al. APOE-related mortality: Effect of dementia, cardiovascular disease and gender. Neurobiol. Aging 30, 1545–1551 (2009).
Deelen, J. et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 10, 1–14 (2019).
Ben-Shlomo, Y. & Marmot, M. G. Survival and cause of death in a cohort of patients with parkinsonism: Possible clues to aetiology? J. Neurol. Neurosurg. Psychiatry 58, 293–299 (1995).
Bennett, D. A. et al. Prevalence of Parkinsonian signs and associated mortality in a community population of older people. N. Engl. J. Med. 334, 71–76 (1996).
Matinolli, M., Korpelainen, J. T., Sotaniemi, K. A., Myllylä, V. V. & Korpelainen, R. Recurrent falls and mortality in Parkinson’s disease: A prospective two-year follow-up study. Acta Neurol. Scand. 123, 193–200 (2011).
Tsuang, D. et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70, 223–228 (2013).
Zhao, N. et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 12, 1809 (2020).
Pu, J. L. et al. Apolipoprotein E genotype contributes to motor progression in Parkinson’s disease. Mov. Disord. 1–6 https://doi.org/10.1002/mds.28805 (2021).
Williams-Gray, C. H. et al. Apolipoprotein e genotype as a risk factor for susceptibility to and dementia in Parkinson’s Disease. J. Neurol. 256, 493–498 (2009).
de Lau, L. M. L., Schipper, C. M. A., Hofman, A., Koudstaal, P. J. & Breteler, M. M. B. Prognosis of Parkinson disease. Arch. Neurol. 62, 1265 (2005).
Castilla-Cortázar, I., Aguirre, G. A., Femat-Roldán, G., Martín-Estal, I. & Espinosa, L. Is insulin-like growth factor-1 involved in Parkinson’s disease development? J. Transl. Med. 18, 1–17 (2020).
Pristerà, A. et al. Dopamine neuron-derived IGF-1 controls dopamine neuron firing, skill learning, and exploration. Proc. Natl. Acad. Sci. USA. 116, 3817–3826 (2019).
Lawton, M. et al. Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J. Neurol. Neurosurg. Psychiatry 89, 1279–1288 (2018).
Lewis, S. J. G. et al. Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J Neurol. Neurosurg. Psychiatry 76, 343–348 (2005).
Iwaki, H. et al. Genetic risk of Parkinson disease and progression: An analysis of 13 longitudinal cohorts. Neurol. Genet 5, 1–14 (2019).
Saunders-Pullman, R. et al. Progression in the LRRK2-Asssociated Parkinson disease population. JAMA Neurol 75, 312–319 (2018).
Sanofi. Sanofi delivered close to double-digit Q4 2020 business EPS(1) growth at CER. https://www.sanofi.com/en/media-room/press-releases/2021/2021-02-05-06-30-00-2170436 (2021).
Knipe, M. D. W., Wickremaratchi, M. M., Wyatt-Haines, E., Morris, H. R. & Ben-Shlomo, Y. Quality of life in young- compared with late-onset Parkinson’s disease. Mov. Disord. 26, 2011–2018 (2011).
Hoehn, M. M., Yahr, M. D., Hoehn, M. M. & Yahr, M. D. Parkinsonism: onset, progression, and mortality. Neurology 17, 427–442 (1967).
Roos, R. A. C., Jongen, J. C. F. & Van Der Velde, E. A. Clinical course of patients with idiopathic Parkinson’s disease. Mov. Disord. 11, 236–242 (1996).
Goetz, C. G., Stebbins, G. T. & Blasucci, L. M. Differential progression of motor impairment in levodopa-treated Parkinson’s disease. Mov. Disord. 15, 479–484 (2000).
Evans, J. R. et al. The natural history of treated Parkinson’s disease in an incident, community based cohort. J. Neurol. Neurosurg. Psychiatry 82, 1112–1118 (2011).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Kichaev, G. et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 10, 1–16 (2014).
Kichaev, G. & Pasaniuc, B. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet. 97, 260–271 (2015).
Kichaev, G. et al. Improved methods for multi-trait fine mapping of pleiotropic risk loci. Bioinformatics 33, 248–255 (2017).
Malek, N. et al. Tracking Parkinson’s: Study Design and Baseline Patient Data. J. Parkinsons. Dis. 5, 947–959 (2015).
Szewczyk-Krolikowski, K. et al. The influence of age and gender on motor and non-motor features of early Parkinson’s disease: Initial findings from the Oxford Parkinson Disease Center (OPDC) discovery cohort. Park. Relat. Disord. 20, 99–105 (2014).
Marek, K. et al. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Wickremaratchi, M. M. et al. Prevalence and age of onset of Parkinson’s disease in Cardiff: a community based cross sectional study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 80, 805–807 (2009).
Foltynie, T., Brayne, C. E. G., Robbins, T. W. & Barker, R. A. The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain 127, 550–560 (2004).
Williams-Gray, C. H. et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 84, 1258–1264 (2013).
Williams-Gray, C. H. et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 132, 2958–2969 (2009).
Corvol, J. C. et al. Longitudinal analysis of impulse control disorders in Parkinson disease. Neurology 91, e189–e201 (2018).
Hustad, E., Myklebust, T. Å., Gulati, S. & Aasly, J. O. Increased Mortality in Young-Onset Parkinson’s Disease The question of prognosis in terms of progression of motor. J. Mov. Disord. 14, 214–220 (2021).
Pihlstrøm, L., Morset, K. R., Grimstad, E. & Vitelli, V. A cumulative genetic risk score predicts motor progression in Parkinson’s disease. Mov. Disord. 31, 487–490 (2016).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Wallace, C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet 17, 1–11 (2021).
Reynolds, R. H. RHReynolds/colochelpR: v0.99.1. https://doi.org/10.5281/ZENODO.5011869 (2021).
Wang, D. et al. Comprehensive functional genomic resource and integrative model for the human brain. Science (80-) 362, 1–13 (2018).
Krohn, L. et al. Genome-wide association study of REM sleep behavior disorder identifies polygenic risk and brain expression effects. Nat. Commun. 13, 1–16 (2022).
Krohn, L. et al. Genome-wide association study of REM sleep behavior disorder identifies novel loci with distinct polygenic and brain expression effects. medRxiv https://doi.org/10.1101/2021.09.08.21254232 (2021).
McDonald, M. L. N. et al. Novel genetic loci associated with osteoarthritis in multi-ancestry analyses in the Million Veteran Program and UK Biobank. Nat. Genet. 54, 1816–1826 (2022).
Acknowledgements
Funding sources: Parkinson’s UK, Aligning Science Across Parkinson’s [ASAP-000478] through the Michael J Fox Foundation for Parkinson’s Research, Southern and Eastern Norway Regional Health Authority. Both the Oxford Discovery (Grant reference J-1403) and Tracking Parkinson’s (PRoBaND) cohorts (grant reference J-1101) were funded by Parkinson’s UK. Oxford Discovery was funded by the Oxford Biomedical Research Centre (BRC). This research was funded in whole or in part by Aligning Science Across Parkinson’s [Grant number: ASAP-000478] through the Michael J. Fox Foundation for Parkinson’s Research (MJFF). For the purpose of open access, the author has applied a CC BY public copyright license to all Author Accepted Manuscripts arising from this submission. This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; project ZIA AG000949. The CamPaIGN study and Cambridge PD Research Clinic were supported by the NIHR Cambridge Biomedical Research Centre (NIHR 203312). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. The CamPaIGN study also received funding from the Wellcome Trust, the Medical Research Council, and the Patrick Berthoud Trust. This research has been conducted using the UK Biobank Resource under Application Number 46450. Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (https://www.ppmi-info.org/accessdata-specimens/download-data). For up-to-date information on the study, visit www.ppmi-info.org. PPMI – a public-private partnership – is funded by The Michael J. Fox Foundation for Parkinson’s Research and funding partners, including 4D Pharma, AbbVie Inc., AcureX Therapeutics, Allergan, Amathus Therapeutics, Aligning Science Across Parkinson’s (ASAP), Avid Radiopharmaceuticals, Bial Biotech, Biogen, BioLegend, BlueRock Therapeutics, Bristol Myers Squibb, Calico Life Sciences LLC, Celgene Corporation, DaCapo Brainscience, Denali Therapeutics, The Edmond J. Safra Foundation, Eli Lilly and Company, Gain Therapeutics, GE Healthcare, GlaxoSmithKline, Golub Capital, Handl Therapeutics, Insitro, Janssen Pharmaceuticals, Lundbeck, Merck & Co., Inc., Meso Scale Diagnostics, LLC, Neurocrine Biosciences, Pfizer Inc., Piramal Imaging, Prevail Therapeutics, F. Hoffmann-La Roche Ltd and its affiliated company Genentech Inc., Sanofi Genzyme, Servier, Takeda Pharmaceutical Company, Teva Neuroscience, Inc., UCB, Vanqua Bio, Verily Life Sciences, Voyager Therapeutics, Inc., and Yumanity Therapeutics, Inc. (https://www.ppmi-info.org/about-ppmi/who-we-are/study-sponsors).
Author information
Authors and Affiliations
Contributions
Concept and design: M.M.X.T., E.J., H.R.M., D.G.G., M.S. Acquisition, analysis or interpretation of data: M.M.X.T., M.A.L., M.I.P., E.B., R.R., A.M.C., S.B., R.H.R., H.I., C.B., S.K., L.H., N.M., K.A.G., N.B., R.A.B., D.J.B., C.B., T.F., N.W.W., C.H.W.-G., O.A.A., M.T., A.E., F.A., A.B., J.-C.C., J.A., M.J.F., M.A.N., A.B.S., N.M.W., Y.B.-S., J.H., M.T.M.H., D.G.G., L.P., H.R.M. Drafting of the manuscript: M.M.X.T., H.R.M. Critical revision of the manuscript: M.M.X.T., M.A.L., R.R., A.M.C., S.B., E.J., H.I., C.B., K.A.G., N.B., R.A.B., D.J.B., T.F., N.W.W., C.H.W.-G., O.A.A., M.T., A.E., A.B., J.-C.C., M.J.F., M.A.N., A.B.S., N.M.W., Y.B.-S., J.H., M.T.M.H., D.G.G., M.S., L.P., H.R.M. Statistical analysis and supervision: M.M.X.T., M.A.L., H.I., Y.B.-S., M.S.
Corresponding authors
Ethics declarations
Competing interests
MMXT is employed by Oslo University Hospital. She has received grant support from Parkinson’s UK, the Michael J Fox Foundation, and South-Eastern Norway Regional Health Authority (Helse Sør-Øst). MAL received fees for advising on a secondary analysis of an RCT sponsored by North Bristol NHS trust. KG is an independent medical consultant and receives payments from the University of Glasgow. She has no other fees or honoraria to report. J-CC has served on advisory boards for Biogen, Denali, Idorsia, Prevail Therapeutic, Servier, Theranexus, UCB; and received grants from Sanofi and the Michael J Fox Foundation outside of this work. RHR is currently employed by CoSyne Therapeutics (Lead Computational Biologist). All work performed for this publication was performed in her own time, and not as a part of her duties as an employee. MAN’s participation in this project was part of a competitive contract awarded to DataTecnica LLC by the National Institutes of Health to support open science research. MAN also currently serves on the scientific advisory board for Character Bio Inc and is a scientific founder at Neuron23 Inc and owns stock. ABS is Associate Editor of npj Parkinson’s Disease. ABS was not involved in the journal’s review of, or decisions related to, this manuscript. OAA is a consultant to HealthLytix. CWG is employed by the University of Cambridge and supported by the Medical Research Council (MR/R007446/1 and MR/W029235/1) and the NIHR Cambridge Biomedical Research Centre (NIHR 203312). She has received research funding from the Cambridge Centre for Parkinson-Plus, Cure Parkinson’s, Parkinson’s UK, The Evelyn Trust, The Rosetrees Trust, Addenbrooke’s Charitable Trust and the Michael J Fox Foundation; speaker payments from GSK and World Parkinson’s Coalition; and consulting fees from Evidera Inc. RAB has served as an advisor to UCB; BlueRock Therapeutics; Novo Nordisk; Aspen Neuroscience, UCB and Transine Therapeutics. He has received lecture fees from Novo Nordisk. He has received grant support from the MRC, Wellcome, Parkinson’s UK, Cure Parkinson’s Trust, EU, Novo Nordisk, DRI, and ASAP. This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. He has received royalties from Wiley and Springer-Nature. DGG is an employee of the University of Glasgow. In the past 12 months, he reports consultancy fees from the Glasgow Memory Clinic; honoraria for chairing or attending meetings from AbbVie and BIAL Pharma. LP has received grant support from the National Health Association, Norway, the Michael J Fox Foundation, and South-Eastern Norway Regional Health Authority (Helse Sør-Øst). HRM is employed by UCL. In the last 12 months, he reports paid consultancy from Roche and Amylyx; lecture fees/honoraria - BMJ, Kyowa Kirin, Movement Disorders Society. Research Grants from Parkinson’s UK, Cure Parkinson’s Trust, PSP Association, Medical Research Council, Michael J Fox Foundation. HRM is a co-applicant on a patent application related to C9ORF72 - Method for diagnosing a neurodegenerative disease (PCT/GB2012/052140).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, M.M.X., Lawton, M.A., Pollard, M.I. et al. Genome-wide determinants of mortality and motor progression in Parkinson’s disease. npj Parkinsons Dis. 10, 113 (2024). https://doi.org/10.1038/s41531-024-00729-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-024-00729-8
This article is cited by
-
A community-led initiative to de-risk and advance Parkinson’s disease therapeutic targets
npj Parkinson's Disease (2025)
