
Abstract
Global restoration and conservation of freshwater biodiversity are represented in practice by works such as the Klamath River Renewal Project (KRRP), the largest dam removal and river restoration in the United States, which has reconnected 640 river kilometers. With dam removals, many biological outcomes remain understudied due to a lack of pre-impact data and complex ecosystem recovery timeframes. To avoid this, we created the KRRP molecular library, an environmental specimen bank, for long-term curation of environmental nucleic acids collected from the restoration project. We used these initial samples, environmental DNA metabarcoding, and generalized linear mixed-effects models to evaluate patterns of pre-dam removal fish richness and diversity. Demonstrating the suitability to resolve biological differences, the baseline shows that tributary and mainstem streams had greater native fish diversity and 2.3–10.7 times greater native fish species richness than reservoirs. These and future sampling efforts should, at a minimum, allow tracking of fish community response to ecosystem restoration. Anticipating the acceleration of omics innovation, we preserved samples for long-term storage and identified requisite phases for sustained function and adaptation of the molecular library: securing a physical storage facility for genetic material, establishing a governance structure, and confirming support for archive management.
Similar content being viewed by others

Introduction
Freshwater habitats are vital for human and ecosystem wellbeing1. Lotic and lentic habitats support economic, recreational, and cultural services, while being home to much larger levels of endemism, fish species richness, and biodiversity than their spatial footprint alone would suggest2. For millennia, human dependence on river ecosystems has created cumulative impacts that have become larger, more widespread, and difficult to manage. This has contributed to a global increase in species extinctions, decline in freshwater species abundance, and a loss of biodiversity and ecosystem function3,4. A rising awareness of the threats and peril to rivers and the human populations served by these ecosystems has emerged2,5,6 and led to calls for management plans addressing restoration and recovery of freshwater ecosystems1,7,8,9,10. These discussions about the conservation and restoration of freshwater biodiversity often recommend measures to maintain or restore river connectivity and the associated ecological processes and functions (e.g., natural flow, sediment, and temperature regimes) contained in free-flowing rivers11,12,13,14. Over the past two decades, dam removal has been increasingly used as a tool to restore river connectivity, while also removing obsolete, unsafe, or inconsequential structures no longer meeting their intended purpose15,16,17. Although most dam removal outcomes are unstudied18, the number and diversity of studies addressing the physical and biological outcomes is increasing19. Yet, there remains ample space to explore new approaches and emerging technologies to address the outcomes from dam removal.
Ecosystems are inherently complex, with multiple interacting species, processes, and environmental factors20. Additionally, baseline conditions may shift over time due to natural variability or human influence, making it difficult to interpret monitoring metrics or determine progress towards recovery goals. Ecosystem changes occur over various temporal scales, from short-term fluctuations to long-term trends. Spatial heterogeneity within and among ecosystems may also influence how monitoring metrics change21. The methods used to assess ecosystem recovery, such as remote sensing, field surveys, and genetic analyses, each have limitations. These might include resolution constraints, scale limitations, and detection limits22,23. Combining multiple methods often provides a more comprehensive view needed for evaluation, but integrating different types of data or consolidating different data systems introduces challenges. The type of monitoring conducted must capture the appropriate scales required for program objectives, including where and when changes are occurring, what is changing, and what is causing change. Identifying appropriate measurable indicators that are informative about ecosystem status and recovery is challenging. Indicators must be sensitive to change, relevant across different scales, and robust to natural variability. Further, a combination of biotic and abiotic indicators is needed, each having specialized requirements for measurement and interpretation24.
Advances in quantitative polymerase chain reaction (qPCR) and next-generation sequencing (NGS) have enabled researchers to ask broad and targeted ecological questions using environmental DNA (eDNA) and environmental RNA (eRNA)25,26,27,28. Environmental DNA and eRNA, collectively referred to as environmental nucleic acids (eNA), are cost effective29, non-invasive30, and effective tools for monitoring the distribution of aquatic species at broad geographic scales31,32. Increasingly, molecular tools including eNA have been used to document broadscale changes to biodiversity after restoration including landscape-scale species reestablishment programs33, stream restoration, changes to land use and watershed management34, and following large-scale dam removals35,36,37. Additionally, the rapid evolution of NGS tools including environmental metagenomics (the collection of genomes in an environmental sample) and environmental metatranscriptomics (the collection of gene transcripts in an environmental sample) presents opportunities to assess community level changes to genetic diversity and gene expression following restoration, particularly with microorganisms38,39.
Environmental specimen banks are programs that curate physical storage of environmental samples through time for future monitoring and research. Although most focus on preserving samples for the analysis of environmental contaminants, chemical trends, human and animal tissues, plant tissues, and environmental samples, molecular focused environmental specimen banks present an opportunity to assess long-term changes in biodiversity from preserved genetic material40. Recently, existing environmental samples stored in environmental specimen banks have been opportunistically utilized to measure changes in biodiversity through time with eNA41,42,43, but challenges remain regarding the ability of regional environmental specimen banks to capture and preserve eNA with sufficient resolution to assess restoration projects44,45,46. It has been shown that cryopreservation of both DNA and RNA in liquid nitrogen successfully preserves and maintains the integrity of nucleic acids over decades. One study reported that RNA isolated from breast cancer samples stored for a minimum of 10 years in the vapor phase of liquid nitrogen exhibited significantly higher RNA Integrity Number equivalent (RINe) values compared to those stored at − 80 °C, indicating superior preservation of RNA quality with liquid nitrogen storage47. Although preserved environmental samples can retain usable genetic material for decades, extracted and purified DNA and RNA can retain sufficient quantity and quality for much longer, possibly up to tens of thousands of years48.
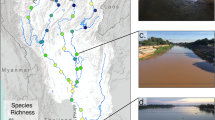
The Klamath River Renewal Project (KRRP; also called Lower Klamath Project) is the largest dam removal and river restoration in the history of the United States and represents a unique opportunity to study landscape-scale change. Extending across northern California and southern Oregon, the project includes the removal of four hydroelectric dams and their associated infrastructure along the Klamath River (Fig. 1). Dam removal and subsequent restoration of the former reservoir footprints to a riverine condition is intended to reconnect over 640 km of habitat for anadromous and migratory fishes, restore native vegetation across over 800 ha of previously drowned land, and improve water quality and habitat conditions within the 305 km of mainstem river downstream from the dams49. Dam removal was completed—with volitional fish passage restored in fall 2024 (Fig. 2)—with anticipated fisheries and ecosystem function benefitting local communities, including members of the Indigenous Tribes who have relied on a healthy and well managed Klamath River since time immemorial50,51,52.

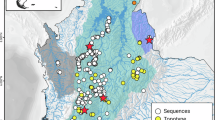
Map of the Klamath River Renewal Project (Lower Klamath Project) and molecular library monitoring sites, including geographic ___location of the Klamath River Basin, the project area including dams removed in 2024, and the Scott River reference sites (upper left), the distribution of the reference sites (upper right), and the mainstem, tributary, and control sites in relation to the reservoir footprints (lower).

Images of before and after dam removal on the Klamath River. (A1) Upstream view of the Iron Gate Reservoir below Fall Creek, prior to the start of reservoir drawdown and dam removal, beginning of January 2024 (Photo credit: resource environmental solutions (RES)). (A2) Same ___location as (A1), now an upstream view of the Klamath River below Fall Creek, taken within the former Iron Gate Reservoir footprint, May 2024 (Photo credit: RES). (B1) Drone image of the Copco 1 Dam, completed in 1918, along the Klamath River during pre-dam removal activities in September 2023 (Photo credit: RES). (B2) Drone image of the Klamath River at the former Copco 1 Dam site following the completion of dam removal activities in October 2024 (Photo credit: RES).
Despite the expected benefits of dam removal, there are few long-term studies that have demonstrated population level responses in fish. The scale and importance of the KRRP presents a unique opportunity to address the long-term outcomes of dam removal on fish population response and the processes involved in ecosystem recovery. We created the Molecular Library (ML) as an environmental specimen bank specifically to preserve eNA as a data legacy for long-term assessment of dam removal outcomes and associated reestablishment of native species along the Klamath River, as well as the global effort to understand biodiversity response to landscape-scale restoration. The ML is a novel approach: to develop an environmental specimen bank that combines the best practices for evaluating biodiversity response to restoration through time53 and accounts for challenges faced by existing environmental specimen banks for the effective capture and preservation of eNA44. Our objectives were to: (1) introduce and document a framework for landscape-scale restoration research and monitoring with eNA that aims to apply best modern practices to create a molecular library of samples for the purpose of short-term use (contemporary analysis) and long-term use (archiving for posterity); and (2) demonstrate sample validation and baseline conditions (pre-dam removal) of fish communities across longitudinally placed sample sites upstream and downstream of the former dams, within reservoir reaches, and reference reaches in tributaries.
Methods
Site selection
We established an initial experimental design anticipated to address both short- and long-term changes within 114 km of river and tributary areas impacted by the KRRP. A key aspect of the design was to ensure that sites would be accessible both physically (regardless of environmental conditions) and jurisdictionally via stable public lands54. Based on previous dam removal monitoring efforts55,56,57, we expected significant short-term changes to the river ecosystem to be encompassed within the project area proximate to the four dams (i.e., spanning from just upstream of the J.C. Boyle uppermost reservoir to 3.5 km downstream of the lowermost Iron Gate Reservoir). As such, we systematically established 20 monitoring locations every 2 km along the Klamath River mainstem and reservoirs and 17 locations every 1 km along selected tributaries within the anadromous zone (Fig. 1). Scaled systematic distance between monitoring locations of 2 km and 1 km respectively were used to reduce the probability of extra-organismal eNA being transported between sites58. However, we also recognize that flexibility of the design for future expansion of sampling locations may be necessary to evaluate long-term outcomes or related study questions54.
We also established two different types of comparison sites that were not directly impacted by dam removal. We selected two “control” sites upstream of anadromous barriers in tributaries of the study reach (Fig. 1). These were intended to isolate the effects of dam removal (i.e., habitat transition from lentic to lotic, restoring natural flow, sediment, and temperature regimes, and upstream passage of aquatic organisms59) from localized natural variability. Yet, tributary sites upstream of barriers tend to be smaller order watersheds that might not be representative of changes experienced within a larger system like the Klamath River. Thus, we added six additional “reference” sites in the Scott River basin, a large tributary downstream of the project area. Having a range of non-treatment reaches from which to compare treatment (i.e., dam removal) effects will be more representative of recovered conditions and biological communities60. We monumented all sites with GPS ___location, documented each site with photographs, and stored site level information in ArcGIS online for coordination between field teams and long-term data integrity and storage.
Field sampling
We collected 405 samples from 44 monitoring locations on 17–20 July, 2023. The summer season from July through August is the preferred time to complete annual sampling due to lower stream discharge that minimizes the dilution of eNA in streams61, increasing the probability of detecting key indicator species including bacteria, algae, macroinvertebrates, amphibians, fishes, and pathogens. The sampling window also overlaps with the juvenile life-stage of native migratory fishes within the watershed, increasing the likelihood that eDNA is captured from this life stage specifically62,63,64. Access allowing, we collected 3 L of stream water from each bank and the center of the channel at each site, for a total of 9 L. The 9 L of water were combined into a single vessel, agitated to encourage homogenization, and decanted into nine replicate samples via a filtering manifold. Each replicate was filtered in the field through 0.45 µm pore-size PVDF Sterivex filters (MilliporeSigma, Burlington, MA, USA; cat #SVHVL10RC), which capture eNA from the environment by trapping particles within the filter matrix, using Masterflex Easy-Load II peristaltic pumps (VWR, Radnor, PA, USA; cat #MFLX77200-52) and sterile Masterflex tubing (VWR, Radnor, PA, USA; cat# MFLX-06509-24) for each monitoring site. Pumps were affixed in parallel to allow for simultaneous filtration of three filter replicates and powered using a brushless, cordless drill with a 12.7 mm spade bit attachment. Field crews followed protocols to assess and minimize the risk of contamination including using sterile single-use filters, caps, and tubing, changing nitrile gloves frequently, and collecting field controls at the start of each sampling day (Fig. 3). Site-level water quality measurements (water temperature, dissolved oxygen, and specific conductance) were collected with a Multiparameter Digital Water Quality Meter (Yellow Springs Instruments, Ohio, USA; model #626870-1) and air temperature with a rotating-vane thermistor (KESTREL 3000—Wind Meter, USA, model #0830). When field sampling was complete, samples were preserved by pipetting into each filter cartridge 1.5 mL of RNAprotect Tissue Reagent (QIAGEN, Hilden, Germany; cat #76106) following practices to maximize the probability of stabilizing genetic material27. Samples were then stored in a portable cooler with blue ice packs before being transferred to a non-frost-free freezer and stored at − 20 °C.

Diagram showing (A) water sample collection and filtration protocol with potential analyses and inference possible over the life of the molecular library eNA archive (including a proportion of samples preserved for eDNA analysis pathways symbolized by the double helix icon and a proportion of samples preserved for eRNA analysis pathways symbolized by the single stranded icon), (B) the proposed sampling timeframe and data purpose over timescales relevant to management and research and as a time capsule, to capture ecosystem responses to dam removal, and (C) status of requisite steps for the establishment of molecular library as an environmental specimen bank, including 1those completed at time of publication and 2those identified as potential future activities.
Molecular methods
Total DNA was isolated from selected filters and purified to remove non-target cellular and environmental contaminants using the QIAamp DNA mini kit (QIAGEN, Hilden, Germany; cat #51306) and following a standard protocol with modifications65. First, we removed RNAprotect Tissue Reagent from each filter by manually shaking the liquid from the cartridge and sterilized the exterior of each filter with a PCR clean wipe (Thomas Scientific, Swedesboro, NJ, USA; cat #C791Q58) to avoid cross contamination. We added 440 μL of the Buffer PBS/Buffer AL/Proteinase K lysis solution65 to each filter by injecting the solution into the Sterivex cartridge using a filtered pipette tip. The filters were incubated for 5 min at 56 °C, then affixed to a Vortex-Genie 2 mixer (Scientific Industries, Bohemia, NY, USA; cat #SI0236) to undergo two 10-min room temperature vortex sessions. Between sessions, the filters were rotated 180° to ensure full coverage of the filter membrane. We used a 3 mL luer lock syringe (Fisher Scientific, Waltham, MA, USA; cat #14-823-435) to transfer the solution from the Sterivex cartridge to a 1.5 mL microcentrifuge tube containing 200 μL of lab-grade ethanol. DNA was then bound using QIAamp mini spin columns, and the remaining purification and elution steps were carried out according to the published protocol65. We processed extraction controls, created by adding 880 μL of the lysis solution to a sterile Sterivex filter, in parallel with samples to confirm sample integrity throughout the extraction procedure. All samples and controls were passed through the Zymo OneStep PCR Inhibitor Removal Kit (Zymo Research, Irvine, California, USA; cat #D6030) following manufacturers guidelines. The DNA extraction steps were completed in a separate pre-PCR space using sterilized surfaces and equipment.
Purified eNA can be analyzed using a variety of molecular techniques. For this study, we used DNA metabarcoding to assess the community-level composition of fish taxa at each sampling ___location. Metabarcoding employs next-generation sequencing with universal primers to sequence a diagnostic region of DNA that allows for species identification across taxa. We used a multiplex of the MiFish-U primer set66 and a modified version of the MiFish-U-F primer (GIQHerp-F), designed to enhance detection of herptile taxa, to sequence a 170 bp region of vertebrate 12S rRNA mitochondrial genome using three-step PCR approach adapted from previously published library preparation methodologies66,67. The initial PCR was completed using non-indexed primers to enrich subsequent reactions for target DNA. Each sample was amplified in triplicate, in a total reaction volume of 10 μL containing 4 μL extracted eDNA, 0.4 μM of each forward primer (MiFish-U-F: 5′-GTCGGTAAAACTCGTGCCAGC-3′, GIQHerp-F: 5′-GCCGGCTAATCTGGTGCCAGC-3′), 0.8 μM MiFish-U-R (5′-CATAGTGGGGTATCTAATCCCAGTTTG-3′), and 1× Qiagen Plus Multiplex Master Mix (QIAGEN, Hilden, Germany; cat #206145). Cycling began with an initial denaturation at 95 °C for 5 min, followed by 35 cycles of 95 °C for 15 s, 5% ramp down to 55 °C for 30 s, and 72 °C for 30 s. The triplicate PCR products were pooled then diluted 1:10 prior to starting the Illumina adapter and barcoding processes.
The Illumina hanging tail adapters were incorporated using the MiFish-U and GIQHerp primer multiplex containing the 33 or 34 bp 5′ Illumina hanging tail adaptor sequences to provide a priming site for the addition of dual indexed barcode sequences. Each reaction consisted of a 12 μL total volume containing 2 μL pooled and diluted product from the previous PCR, 0.3 μM of each Illumina adapter forward primer, 0.6 μM of the Illumina adapter reverse primer, and 6 μL KAPA HiFi HotStart ReadyMix (Roche Diagnostics, Indianapolis, IN; cat #07958935001). The cycling profile was as follows: 95 °C for 5 min, 5 cycles of 98 °C for 20 s, 1% ramp down to 65 °C for 15 s, and 72 °C for 15 s, then 7 cycles of 98 °C for 20 s, 5% ramp down to 65 °C for 15 s, 72 °C for 15 s. PCR products were diluted 1:10 and used as template in the final PCR step. The paired-end dual indices that allow for sample identification and de-multiplexing were incorporated during the final PCR step. Each PCR was completed in a total volume of 12 μL, composed of 0.3 μM of the forward and reverse index primers, 6 μL 1× KAPA HiFi HotStart ReadyMix, and 1 μL of the diluted product from the previous PCR. Amplification started with 95 °C for 3 min, followed by 10 cycles of 98 °C for 20 s, 5% ramp down to 72 °C for 15 s, and final extension 72 °C for 5 min. All PCR steps were completed using BioRad C1000 Touch thermal cyclers (Bio-Rad Laboratories, Hercules, CA, USA) in a designated PCR space.
Equal volumes of the indexed PCR products were pooled, then size selected (c. 370) using 2% gel electrophoresis and purified using QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany; cat #28704) following the manufacturers guidelines for next-generation sequencing. Purified libraries were quantified using the Qubit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA; cat #Q33226) and Qubit dsDNA HS assay Kit (Thermo Fisher Scientific, Waltham, MA, USA; cat # Q33231), and sequenced on the Illumina Miseq system (Illumina, San Diego, CA, USA) using the v2 300-cycle chemistry. The final loading concentration was 8 pM with a 10% PhiX spike-in added as a sequencing control. Using a UV sterilized hood, we prepared master mix for all PCR steps and added extracted DNA during the initial PCR. All intermediate dilution, DNA transfer, and final pooling steps were completed in designated post-PCR spaces using sterilized pipettes and bench tops. No template PCR controls were processed in parallel with samples and sequenced to confirm process integrity.
To determine provisional species identification, the resultant sequencing data were compiled and processed using the MetaWorks pipeline, 12S vertebrate classifier, and default parameters68. The data output from this pipeline grouped exact sequence variants (ESV) into zero-radius operational taxonomic units (ZOTUs), determined the number of times an ESV was observed in each sample (sequence reads), and provided a provisional species identification of each sequence using the selected classifier. We removed any ESVs with less than 100 sequence reads to screen out potential artifact sequences. We applied a conservative screening method to eliminate spurious sequences and focus on those with sufficient abundance to be considered reliable. We recognize that this method may result in loss of real diversity, however this approach has been advised for ecological inference studies with high read depth69. The provisional taxonomic assignment was verified against the NIH National Center for Biotechnology Information (NCBI) GenBank nr reference database (Accessed 20 Dec. 2024) using the Basic Local Alignment Search Tool (BLAST70; https://blast.ncbi.nlm.nih.gov/Blast.cgi). We used the standard nucleotide BLAST (blastn suite) to compare detected sequences to sequences stored in the core nucleotide database (core_nt). The BLAST output was further curated to determine the final species identification. ESVs were assigned to species when sequence identity was greater than or equal to 97% with 100% query coverage to a single species. ESVs that matched multiple species with the same identity percentage and query coverage criteria were further evaluated for historical occurrence in the sampling region, and only species that could occur in the sampling region were assigned. If more than one species matched within the stated BLAST criteria and could co-occur in the sampling region, the ESV was assigned to the taxonomic level that appropriately captured all potential matches (e.g., Cottus spp.). ESVs that matched GenBank sequences with less than 95% identity or failed to produce any matches were considered too dissimilar to be accurately identified and were removed from the analysis. Any detections that could result from anthropogenic inputs (human, cat, dog, cow, chicken, and pig) were removed from analysis. Additionally, we removed detections of fish species that were anomalous for the sampling region and have been previously identified as contaminants. These species included common bleak (Alburnus alburnus), common barbell (Barbus barbus), common chub (Squalius cephalus), and ballyhoo (Hemiramphus brasiliensis).
Statistical methods
We used sequence reads of fish taxa to visualize data and calculate diversity metrics for analysis. The average rarefied sequence read concentration per taxon across three site replicates was calculated based on the volume of water that was filtered (mL), the re-suspended volume of purified DNA from DNA extraction (µL), the volume of purified DNA added to the initial PCR (µL per reaction), and the sequence reads per taxon (reads per reaction). The flow-adjusted sequence read rate per monitoring ___locationi of taxonj (reads per secondi,j) was calculated whereby monitoring ___location specific discharge (Qi) is estimated as a function of contributing drainage area of monitoring ___locationi, and reads per secondi,j is equal to the product of reads per mL taxonj and mL per second (Qi)71. Native fish diversity (diversity) was estimated per monitoring ___location by the Shannon–Wiener diversity index using R-package vegan based on relative abundance (reads per secondi,j)72. Native fish richness (richness) per monitoring ___location was calculated based on the total number of native fishes detected with greater than 100 sequence reads. Site-level species composition relationships were visualized via non-metric multidimensional scaling (NMDS) using a Bray–Curtis dissimilarity matrix generated from rarefied sequence reads. Species vectors based on significant correlations (p < 0.001) with ordination axes were overlayed to highlight species contributing to group separation. All NMDS calculations were completed using the vegan R-package and visualized at the habitat scale (stream vs. reservoir) with 95% confidence ellipses using ggplot2 R-package72. We performed all statistical analyses in R73.
The relationships between the covariates of habitat type (reservoir vs. stream), stream size (mainstem vs. tributary), control versus impact, reference versus impact, dissolved oxygen (mg per L), and specific conductance (µS per cm) and response variables were evaluated using generalized linear mixed-effects models (GLMMs). Diversity was modeled as a binomial response, with extreme low diversity sites (Shannon–Wiener diversity index < 0.05) and normal or high diversity sites (Shannon–Wiener diversity index > 0.05). Richness was modeled as a count. To identify properly specified models, a binomial GLMM (diversity), with a logit link function, and a Poisson GLMM (richness), with a log link function, were fit with a random effect for waterbody and the fixed effects described above. Additional explanatory variables were not considered to avoid multicollinearity which was examined with R-package Highland Statistics Ver. 1074. The Poisson GLMM was assessed for overdispersion via data simulation with the dispersion_check function in R-package inlatools75. Because no evidence of overdispersion was found, other probability density functions were not considered. Continuous explanatory variables were scaled to have a mean of zero and a standard deviation of one.
We used spatial Pearson residuals of non-spatial GLMMs to construct semi-variograms to test for spatial autocorrelation76. Semi-variograms of Euclidean distance on the scale of 5000 m between sites were assessed and the percentage of the residual variance associated with the spatial effect was quantified with the sill to nugget ratio77. The semi-variograms revealed evidence of spatial autocorrelation on the scale of ~ 2500 m for both models. To account for the residual spatial autocorrelation, the GLMMs were fitted with stochastic partial differential equations (SPDEs) to introduce Gaussian spatial random field effects. Models were fitted using the integrated nested Laplace approximation (INLA), a method that uses a Bayesian framework and was implemented in R software76,78,79. Additional information on model parameterization, INLA model fitting, diagnostics, and interpretation of the spatial random field can be found in the Supplemental Materials: Statistical Methods.
We determined model goodness of fit (GOF) via posterior predictive checks by comparing simulated and observed data and summarizing with a Bayesian p value. The GOF of models was considered suitable if the Bayesian value was between 0.1 and 0.9. Additionally, predictive power of the properly specified binomial GLMM was assessed by calculating area under the Receiver Operating Characteristic (ROC) curve (AUC) to measure the model’s true positive rate (sensitivity) with R-ppackage pROC, and the properly specified Poisson GLMM was assessed by calculating the percent of the variance explained by the model with pseudo-R2 (R2)80. Because single global models were fitted for diversity and richness, only those parameters with 95% credible intervals (95% CI) that did not span zero were considered significant and suitable for inference.
Results
We processed three replicated eDNA samples for each of the 44 monitoring locations across the study area in July 2023, placing the remaining six samples per site into the molecular library archive for future use. A total of 132 samples, 3 negative extraction controls, and 6 negative PCR controls were successfully sequenced. The metabarcoding methods and bioinformatic pipeline resulted in a total of 10,682,995 reads (mean = 71,697 ± 50,949 reads/sample). The total number of reads per sample prior to filtering ranged from 30 to 255,711. Following removal of ESVs with less than 100 sequence reads, total read count decreased to 10,634,394 (mean = 77,060 ± 48,536 reads/sample), with a range of 841 to 255,234 reads per sample. Taxonomic identification and subsequent removal of poor matches to GenBank and taxa from anthropogenic sources reduced read count to 10,184,373 total reads (mean = 75,439 ± 46,830 reads/sample) with 780–254,903 reads/sample. Of the remaining sequences, 9,863,134 reads (mean = 76,458 ± 45,364 reads/sample) were identified as originating from fish taxa, with a median number of reads per taxon, pooled over monitoring locations, of 5844. We detected low levels of cross-sample contamination in some extraction controls and PCR controls (mean = 32 ± 154 reads/sample). The extraction and PCR controls were free of contamination following the removal of ESVs with less than 100 reads. Common bleak, common barbell, and common chub were detected in 0.75% of samples and ballyhoo was detected in 9.8% of samples at greater than 100 reads. All environmental samples produced data that passed quality control. However, two of three samples collected at Scotch Creek did not contain fish detections and were therefore not included in subsequent analysis.
We detected 8 native fish taxa and 13 exotic fish taxa across the monitoring locations. In addition to fishes, we detected eDNA from 10 reptile and amphibian taxa, 12 bird taxa, and 14 mammal taxa (Supplemental Materials Table 1-Metabarcoding Species Data). Shannon–Wiener diversity index of native fish species at monitoring locations ranged from 0 to 1.61 with a median value of 0.79. Native fish richness ranged from zero to eight taxa with a median value of four. Data visualization with NMDS revealed a grouping by habitat type (reservoir vs. stream), with all the reservoir monitoring locations falling within the reservoir 95% confidence ellipse, and all but four stream monitoring locations falling within the stream 95% confidence ellipse (Fig. 4). Significant species vectors included the native fishes: Catostomidae, speckled dace (Rhinichthys osculus), and rainbow trout/steelhead (Oncorhynchus mykiss), and the exotic fishes: goldfish (Carassius aurataus), yellow perch (Perca flavescens), golden shiner (Notemigonus crysoleucas), and black crappie (Pomoxis nigromaculatus) (Fig. 4). These results suggest that group differences along the first NMDS axis are largely driven by differences in native and exotic fish species presence.

Non-metric multidimensional scaling (NMDS) plot visualizing site-level species composition based on a Bray–Curtis dissimilarity matrix from rarefied sequence reads. Significant species vectors (p ≤ 0.001) are overlaid to highlight species-ordination relationships (common names), with sites grouped by habitat type (stream vs. reservoir) and 95% confidence ellipses displayed. Corresponding scientific names available in Supplementary Information: Supplemental Table 1—Metabarcoding Species Data.
The GLMM modeling framework resulted in ecological models that assessed the relationship between the covariates of habitat type (reservoir vs. stream), stream size (mainstem river vs. tributary), control versus impact, reference versus impact, dissolved oxygen (mg per L), and specific conductance (µS per cm) and response variables (diversity and richness). The diversity model passed GOF and resulted in a true positive rate of 96.9% (AUC = 0.969). The binomial GLMM had three covariates that did not span zero: habitat type, stream size, control versus impact, and reference versus impact. The model suggests that after accounting for the random effects of waterbody and spatial position, the log-odds that reservoir locations will have extreme low native fish diversity is at least 7.71 times (95% credible interval (CI) 7.71–40.8 times) greater than the log-odds that stream locations will have extreme low native fish diversity. Additionally, the log-odds that mainstem sites had low native fish diversity was lower than that of tributary sites (95% CI − 35.4 to − 3.73), and that impact sites in the dam removal reach had low diversity was lower than the reference sites in the Scott River Watershed (95% CI − 22.6 to − 1.31) (Fig. 5).

Four-panel figure illustrating model results for native fish diversity and richness across habitats. (A) Forest plot showing the effects of habitat type (reservoir vs. stream), stream size (mainstem vs. tributary), control versus impact, reference versus impact, scaled dissolved oxygen (mg/L), and scaled specific conductance (µS/cm) on the odds of non-extreme low native fish diversity (error bars represent 95% credible intervals). (B) Receiver operating characteristic (ROC) curve displaying the area under the curve (AUC) fit (0.91) for the diversity model, indicating a high true positive rate. (C) Forest plot of covariate effects on native species richness rate ratios with 95% credible intervals. (D) Scatter plot of fitted versus observed values, showing model accuracy in predicting native species richness across sites (R2 = 0.64).
The richness model passed GOF and described 71% of the variance in native fish taxa richness (R2 = 0.705) (Fig. 5). The model suggests that after accounting for the random effects of waterbody and spatial position, streams were expected to have 2.3–10.7 (95% CI) times greater native fish richness than reservoirs. Additionally, tributaries were expected to have between 13.8 and 56.3% less (95% CI) native fish richness than mainstem river sites. Model validation plots, model formulas, and model fit summaries are available in the Supplemental Materials: Statistical Methods.
Discussion
We created the KRPP molecular library as a forward thinking, long-term data framework that can fill an important gap in understanding the outcomes of a large-scale dam removal and river restoration. The dam removal literature has a paucity of long-term data showing ecosystem outcomes and complexities of processes that generally have pronounced short-term effects (e.g., restoration of connectivity, transitioning of lotic to lentic conditions) followed by a long-term response59. The molecular library introduces a framework for landscape-scale restoration research and monitoring with eNA. By collecting eNA from the Klamath River and tributaries before dam removal and establishing a repeatable protocol for subsequent collection, preservation, and analysis, we created a baseline for managers and researchers to retrospectively query changes to the pre-impact condition. Additionally, we demonstrated the validity of this methodology for discerning patterns in landscape biodiversity by analyzing the effects of pre-dam removal habitat types on the native fish community. While our data analysis utilized a conservative bioinformatics approach, we have made all generated data publicly available for reanalysis as new questions and bioinformatics technologies arise. This approach supports both transparency and adaptability in the ongoing interpretation of ecological change and ecosystem recovery. Although useful in the current context, we identified three requisite phases of development for the long-term maintenance and function of the molecular library as an eNA environmental specimen bank: (1) a physical facility for the long-term storage of extracted genetic material, (2) the establishment of a formalized governance structure to guide the ethical and equitable use of the finite genetic material, and (3) the identification of consistent support for archive management and the development and curation of a user-friendly, public-facing database of sequencing data.
Utility and design of the molecular library
The molecular library currently consists of 44 monitoring locations, spanning approximately 114 km of the Klamath River and tributaries. The spatial scale used ensures that localized effects are captured alongside broader ecosystem changes. The envisioned multi-decade temporal scale can help distinguish between short-term responses and true recovery trends. Partitioning short- and longer-term effects would not be possible without establishing a stable long-term storage and archive for eNA, in addition to an eNA “time capsule” for posterity.
The molecular library design incorporated monitoring reference and control sites that are unaffected by the dam removal but have similar ecological and environmental conditions. The reference sites will provide information on natural variability, helping to isolate the effects of dam removal from other environmental changes. In the baseline samples, we found evidence that the reference sites in the Scott River Watershed had weak to moderate odds of having lower native fish diversity than similar sites that will be impacted by dam removal. Additionally, we found evidence that tributaries had lower native fish diversity than mainstem Klamath River sites. These results highlight the importance of including pre-impact data and both reference and control sites as well as aquatic habitat strata (i.e., mainstem vs. tributary) when creating a baseline to assess recovery trends53,54.
The molecular library will enable biological (e.g., community composition, relative abundance) and physical (e.g., sediment transport, water quality) metrics to be combined in the assessment. Integrated assessments are preferred, and have been planned for in the design, as integration provides a more holistic view of ecosystem recovery critical to understanding the full impact of dam removal. Additionally, the library has the capacity to support the use of ecological models to study outcomes of dam removal under various scenarios, with a short-term emphasis on biological responses. Supporting ecological models may help to study outcomes, guide adaptive management, and identify key uncertainties where additional data collection may be needed. As exemplified herein, these data are highly suitable for detecting differences in community composition by habitat type and confirmed our pre-dam removal expectations: that the odds of low native fish diversity are far greater among reservoir sites along the Klamath River than in streams, and that streams in the study area have greater native species richness than reservoir sites (Fig. 6).

Heat map of the quantity of flow-corrected DNA sequences detected per taxon at each sampling ___location (blue), native species Shannon–Weiner diversity index (green), and native species richness (grey). Darker colors indicate larger values. The plot panels are split vertically with “Reservoir” locations on the left and “Stream” locations on the right, and “Exotic” taxa detected in the top half and “Native” taxa on the bottom half. Sampling locations are plotted downstream to upstream within their respective panels. Scientific names provided in Supplemental Table 1.
The library can be used to generate information about key indicator species expected to respond strongly to changes in the ecosystem or significant management decisions. Early signs of ecological recovery or deterioration would be seen from response of indicator species, providing timely information about effectiveness of dam removal. Pre-dam removal conditions revealed that reservoirs had low native fish diversity, and they were characterized by the presence of exotic species that were generally absent or in low abundance in streams (Fig. 6). These differences in species composition were visualized using NMDS plots (Fig. 4), that showed a clear differentiation between stream and reservoir groups, which can be tracked through time when comparing post-dam removal species compositions. Additionally, these results are consistent with the known species composition in the basin between lentic and lotic habitats81. The emergence of significantly correlated presence of non-native indicator species within reservoir sites demonstrated the utility of metabarcoding to illuminate differences in species composition and could provide a useful approach in the future to track the ecosystem response trajectory at former reservoir sites. Timely and consistent information about indicator species would allow for adaptive adjustments in monitoring, management, and restoration strategies, ultimately increasing the chances of successful recovery.
Ecological models used to relate eNA concentration in flowing waters to the distribution and abundance of aquatic species should account for directional transport over space and time, dilution, decay, deposition, and entrainment of genetic material, throughout a river network82,83. Additionally, species distribution models should account for the potential for the dispersion of the species that release genetic material to be spatially autocorrelated as well75. However, reservoirs and other impoundments to regular streamflow represent challenges for using existing models that integrate hydrology and the transport of genetic material by violating assumptions of unidirectional flow84,85. Due to a series of reservoirs being present in the pre-dam removal condition on the Klamath River, we chose to use estimated stream discharge as a proxy for downstream transport distance of genetic material58 and use distances between monitoring sites to reduce the probability of extra-organismal eNA being transported between sites. In other river systems where isolation by transport distance is not suitable (i.e., greater spatial sampling frequency is required), or when target organisms may predominantly release organismal-eNA (e.g., spores, gametes, larvae) that persist over longer transport distances86,87, additional sampling considerations and models that incorporate a spatial stream-network (SSN) autocorrelative structure may be warranted. Although eDNA species detections in this study were largely consistent with the known species composition in the basin by habitat type81, due to the proximity of upstream reservoirs to three monitoring sites in the Klamath River downstream of Iron Gate Reservoir, Copco Reservoir, and JC Boyle Reservoir (Fig. 6 ___location numbers four, eight, and eleven respectively), it remains unclear whether the detections at those sites were a function of the downstream transport of eDNA or downstream transport of the species themselves. Future studies hoping to use eNA to describe change in species compositions following large-scale geomorphic and hydraulic changes associated with dam removal, may require accounting for eNA transport dynamics in reservoirs to describe the change from baseline conditions.
Dam removal is expected to alter the connectivity among populations and community networks, which in turn may cause shifts in demography, reproductive success, and life history diversity59,88,89,90. Given that data from the molecular library would provide information on where and when species occur, observed changes to species distributions would support genetic monitoring activities. Genetic data can provide insights into the connectivity of populations, potential re-establishment events, and the overall health of species that might not be apparent from population counts alone91.
Building a tool for the future: roles, governance, and establishing a long-term molecular sample library
The goal to establish a long-term archive of eNA samples to track ecosystem response to the historical KRRP arises from two fundamental realizations. First, genomic technologies, reference libraries, and phylogenetic metaknowledge will continue to advance over time, providing new tools, approaches, and interpretations92 that will be incorporated into molecular ecology applications such as studying the biological and ecological outcomes of dam removal. For example, having contemporaneous samples from the time periods before and immediately following dam removal, as well as future samples that become available, should be useful for future researchers applying these new technologies and asking questions that we currently cannot anticipate. For example, life history diversity of salmonids increased in the Elwha River following dam removal90 and genomic variation corresponded with traditionally observed ecotypes of Pacific lamprey (Entosphenus tridentatus) in the Klamath River93. How expression of life history diversity in the Klamath River Basin unfolds is yet to be seen, but curating collected genetic material and associated metadata, and future genomic applications of eNA, will maximize utility of the molecular library far beyond our immediate use. The second realization is that the response of the ecosystem to dam removal will continue to unfold over the next several decades, a timespan rarely encompassed in dam removal or river restoration evaluations19. Although early results in the Klamath River and elsewhere show that fish readily occupy upstream areas following restored connectivity, documenting how that translates into increased productivity, life history diversity, and community dynamics can take longer to unfold35,88,94,95. For example, the life span of Chinook salmon (Oncorhynchus tshawytscha) and steelhead trout (Oncorhynchus mykiss) dictates that only about two or three generations pass per decade (i.e., spawner-to-spawner), meaning that the cumulative response of populations will unfold over a timespan exceeding typical funding cycles. The long-term archive of eNA will provide an opportunity to study how these ecological processes evolved over several decades, which will improve our understanding of the complexity of river restoration.
Despite the obvious benefits of data and sample archives, the reuse of genetic and genomic datasets is uncommon due to the lack of a formalized structure for sample archiving, discovery, and metadata96. Although a formalized governance structure and permanent ___location for the molecular library has yet to be determined, we propose that it follow the principles outlined in several review papers related to genetic-based environmental/tissue sample archives (Table 1)96,97,98,99,100,101. To ensure that data are discoverable and reusable, adoption of FAIR practices (i.e., findable, accessible, interoperable, and reusable98) into the molecular library data sharing agreements would ensure that sample accession numbers and digital object identifiers of studies could be used to track sample use across projects and maintain interoperability. This formalized structure must ensure that future users are able to discover the archive and assess its ability to meet their needs, establish roles and responsibilities as a molecular library user, and include processes for adding additional samples to the archive, with appropriate metadata and data discoverability. Similarly, a robust metadata requirement is recommended, so the existing and future molecular library samples contain the necessary details (e.g., at nested levels of site, filter, and extracted DNA and RNA) of the study and sample context, which is essential for future use and reuse. Data access protocols, including adherence to data sharing guidelines, must be streamlined so that requests to the library—both for repositing new samples and using existing samples—are dealt with in a transparent manner over reasonable timeframes. Finally, determining a home (or homes) for the library, especially the time capsule element, with appropriate and resilient facilities (i.e., − 80 °C liquid nitrogen storage), is critical for ensuring the long-term viability of the samples and any data that are generated from their use.
Our intention is to curate the molecular library to enhance the ability for Tribal and agency managers and researchers, local communities, academic institutions, and interested parties to study landscape-scale biological response to dam removal and restoration. Continued engagement with these groups and subsequent additions of samples and sequence reads will facilitate the iterative refinement of the molecular library as an equitably governed public data resource and a progressive tool that provides the genetic material for retrospective analyses of previously unstudied dam removal outcomes.
Data availability
All data generated or analyzed during this study are available in publicly accessible repositories. The datasets, along with associated metadata and analysis scripts, are hosted on GitHub and can be accessed at [https://github.com/Dylan-Keel/Klamath-River-Renewal-Project-Molecular-Library] and permanent data hosting repository services are provided by DRYAD: [DOI: https://doi.org/10.5061/dryad.0cfxpnwcn]. This repository contains metadata associated with molecular library samples and sampling locations, complete code for data visualization and analysis, photos, figures, and raw sequence read data ensuring transparency and reproducibility of the study. Sequencing data generated in this study have been deposited in the NCBI BioProject database under accession number PRJNA1236377. Additionally, Supplemental materials outlining additional statistical and molecular methods, as well as considerations for the governance of the molecular library are provided therein.
References
Lynch, A. J. et al. People need freshwater biodiversity. WIREs Water 10, e1633 (2023).
Dudgeon, D. et al. Freshwater biodiversity: Importance, threats, status and conservation challenges. Biol. Rev. 81, 163 (2006).
Jelks, H. L. et al. Conservation status of imperiled North American freshwater and diadromous fishes. Fisheries 33, 372–407 (2008).
Reid, A. J. et al. Emerging threats and persistent conservation challenges for freshwater biodiversity. Biol. Rev. 94, 849–873 (2019).
Vörösmarty, C. J. et al. Global threats to human water security and river biodiversity. Nature 467, 555–561 (2010).
Tickner, D. et al. Bending the curve of global freshwater biodiversity loss—An emergency recovery plan. Bioscience 70, 330–342 (2020).
Maasri, A. et al. A global agenda for advancing freshwater biodiversity research. Ecol. Lett. 25, 255–263 (2022).
Arthington, A. H. et al. Accelerating environmental flow implementation to bend the curve of global freshwater biodiversity loss. Environ. Rev. 32, 387–413 (2023).
Birnie-Gauvin, K. et al. The RACE for freshwater biodiversity: Essential actions to create the social context for meaningful conservation. Conserv. Sci. Pract. 5, e12911 (2023).
Pecl, G. T. et al. Biodiversity redistribution under climate change: Impacts on ecosystems and human well-being. Science 355, eaai9214 (2017).
Poff, N. L. et al. The natural flow regime. Bioscience 47, 769–784 (1997).
Beechie, T. J. et al. Process-based principles for restoring river ecosystems. Bioscience 60, 209–222 (2010).
Silva, A. T. et al. The future of fish passage science, engineering, and practice. Fish Fish. 19, 340–362 (2018).
Thieme, M. et al. Measures to safeguard and restore river connectivity. Environ. Rev. 32, 366–386 (2024).
O’Connor, J. E., Duda, J. J. & Grant, G. E. 1000 dams down and counting. Science 348, 496–497 (2015).
Magilligan, F. J. et al. River restoration by dam removal: Enhancing connectivity at watershed scales. Elem. Sci. Anthr. 4, 000108 (2016).
Duda, J. J. & Bellmore, J. R. Dam removal and river restoration. In Encyclopedia of Inland Waters, 576–585. https://doi.org/10.1016/B978-0-12-819166-8.00101-8 (Elsevier, 2022).
Bellmore, J. R. et al. Status and trends of dam removal research in the United States. WIREs Water 4, e1164 (2017).
McCaffery, R., Duda, J. J., Soissons, L. & Roussel, J.-M. Editorial: Large-scale dam removal and ecosystem restoration. Front. Ecol. Evol. 12, 1471146 (2024).
Riva, F. et al. Toward a cohesive understanding of ecological complexity. Sci. Adv. 9, eabq4207 (2023).
Sparrow, B. D. et al. Effective ecosystem monitoring requires a multi-scaled approach. Biol. Rev. 95, 1706–1719 (2020).
Cavender-Bares, J. et al. Integrating remote sensing with ecology and evolution to advance biodiversity conservation. Nat. Ecol. Evol. 6, 506–519 (2022).
Bernatchez, L., Ferchaud, A.-L., Berger, C. S., Venney, C. J. & Xuereb, A. Genomics for monitoring and understanding species responses to global climate change. Nat. Rev. Genet. 25, 165–183 (2024).
Nicholson, E. et al. Scientific foundations for an ecosystem goal, milestones and indicators for the post-2020 global biodiversity framework. Nat. Ecol. Evol. 5, 1338–1349 (2021).
Clark, D. R. et al. Streams of data from drops of water: 21st century molecular microbial ecology. WIREs Water 5, e1280 (2018).
Levi, T. et al. Environmental DNA for the enumeration and management of Pacific salmon. Mol. Ecol. Resour. 19, 597–608 (2019).
Miyata, K. et al. Comparative environmental RNA and DNA metabarcoding analysis of river algae and arthropods for ecological surveys and water quality assessment. Sci. Rep. 12, 19828 (2022).
Carraro, L., Blackman, R. C. & Altermatt, F. Modelling environmental DNA transport in rivers reveals highly resolved spatio-temporal biodiversity patterns. Sci. Rep. 13, 8854 (2023).
Evans, N. T., Shirey, P. D., Wieringa, J. G., Mahon, A. R. & Lamberti, G. A. Comparative cost and effort of fish distribution detection via environmental DNA analysis and electrofishing. Fisheries 42, 90–99 (2017).
Goldberg, C. S. et al. Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307 (2016).
Sutter, M. & Kinziger, A. P. Rangewide tidewater goby occupancy survey using environmental DNA. Conserv. Genet. 20, 597–613 (2019).
Miya, M. Environmental DNA metabarcoding: A novel method for biodiversity monitoring of marine fish communities. Annu. Rev. Mar. Sci. 14, 161–185 (2022).
Adams, A. J. et al. From eDNA to decisions using a multi-method approach to restoration planning in streams. Sci. Rep. 14, 14335 (2024).
Dyson, K. et al. Coupling remote sensing and eDNA to monitor environmental impact: A pilot to quantify the environmental benefits of sustainable agriculture in the Brazilian Amazon. PLoS ONE 19, e0289437 (2024).
Duda, J. J. et al. Environmental DNA is an effective tool to track recolonizing migratory fish following large-scale dam removal. Environ. DNA 3, 121–141 (2021).
Muha, T. P., Rodriguez-Barreto, D., O’Rorke, R., Garcia De Leaniz, C. & Consuegra, S. Using eDNA metabarcoding to monitor changes in fish community composition after barrier removal. Front. Ecol. Evol. 9, 629217 (2021).
Huang, C. S. et al. Early detection and recovery of river herring spawning habitat use in response to a mainstem dam removal. PLoS ONE 18, e0284561 (2023).
Neufeld, J. D. Migrating SSU rRNA gene surveys to the metagenomics era. Environ. Microbiol. Rep. 9, 23–24 (2017).
Trench-Fiol, S. & Fink, P. Metatranscriptomics from a small aquatic system: Microeukaryotic community functions through the diurnal cycle. Front. Microbiol. 11, 1006 (2020).
Küster, A., Becker, P. R., Kucklick, J. R., Pugh, R. S. & Koschorreck, J. The international environmental specimen banks—Let’s get visible. Environ. Sci. Pollut. Res. 22, 1559–1561 (2015).
Díaz, C. et al. Aquatic suspended particulate matter as source of eDNA for fish metabarcoding. Sci. Rep. 10, 14352 (2020).
Krehenwinkel, H. et al. Environmental DNA from archived leaves reveals widespread temporal turnover and biotic homogenization in forest arthropod communities. Elife 11, e78521 (2022).
Sullivan, A. R., Karlsson, E., Svensson, D. & Brindefalk, B. Airborne eDNA captures three decades of ecosystem biodiversity. bioRxiv (2023).
Zizka, V. M. A., Koschorreck, J., Khan, C. C. & Astrin, J. J. Long-term archival of environmental samples empowers biodiversity monitoring and ecological research. Environ. Sci. Eur. 34, 40 (2022).
Dysthe, J. C. et al. Repurposing environmental DNA samples—detecting the western pearlshell (Margaritifera falcata) as a proof of concept. Ecol. Evol. 8, 2659–2670 (2018).
Jarman, S. N., Berry, O. & Bunce, M. The value of environmental DNA biobanking for long-term biomonitoring. Nat. Ecol. Evol. 2, 1192–1193 (2018).
Auer, H. et al. The effects of frozen tissue storage conditions on the integrity of RNA and protein. Biotech. Histochem. 89, 518–528 (2014).
Coudy, D., Colotte, M., Luis, A., Tuffet, S. & Bonnet, J. Long term conservation of DNA at ambient temperature. Implications for DNA data storage. PLoS ONE 16, e0259868 (2021).
U.S. Department of the Interior, Bureau of Reclamation, Klamath Basin Area Office, Mid Pacific Region. Final Biological Assessment, the Effects of the Proposed Action to Operate the Klamath Project from April 1, 2013 Through March 31, 2023 on Federally-Listed Threatened and Endangered Species (2012).
Anderson, M. K. & Lake, F. K. California Indian ethnomycology and associated forest management. J. Ethnobiol. 33, 33–85 (2013).
Lake, F. K. et al. Returning fire to the land: Celebrating traditional knowledge and fire. J. For. 115, 343–353 (2017).
Marks-Block, T., Lake, F. K. & Curran, L. M. Effects of understory fire management treatments on California Hazelnut, an ecocultural resource of the Karuk and Yurok Indians in the Pacific Northwest. For. Ecol. Manag. 450, 117517 (2019).
England, J. et al. Best practices for monitoring and assessing the ecological response to river restoration. Water 13, 3352 (2021).
Lindenmayer, D. B. & Likens, G. E. The science and application of ecological monitoring. Biol. Conserv. 143, 1317–1328 (2010).
Hart, D. D. et al. Dam removal: Challenges and opportunities for ecological research and river restoration. Bioscience 52, 669 (2002).
Doyle, M. W. et al. Stream ecosystem response to small dam removal: Lessons from the Heartland. Geomorphology 71, 227–244 (2005).
Foley, M. M. et al. Dam removal: Listening in. Water Resour. Res. 53, 5229–5246 (2017).
Jo, T. & Yamanaka, H. Meta-analyses of environmental DNA downstream transport and deposition in relation to hydrogeography in riverine environments. Freshw. Biol. 67, 1333–1343 (2022).
Bellmore, J. R. et al. Conceptualizing ecological responses to dam removal: If you remove it, what’s to come?. Bioscience 69, 26–39 (2019).
Palmer, M. A. et al. Standards for ecologically successful river restoration. J. Appl. Ecol. 42, 208–217 (2005).
Curtis, A. N., Tiemann, J. S., Douglass, S. A., Davis, M. A. & Larson, E. R. High stream flows dilute environmental DNA (eDNA) concentrations and reduce detectability. Divers. Distrib. 27, 1918–1931 (2021).
Thompson, N. F. et al. A complex phenotype in salmon controlled by a simple change in migratory timing. Science 370, 609–613 (2020).
Hamilton, J. B., Curtis, G. L., Snedaker, S. M. & White, D. K. Distribution of anadromous fishes in the Upper Klamath River Watershed prior to hydropower dams—A synthesis of the historical evidence. Fisheries 30, 10–20 (2005).
Sutton, R. & Soto, T. Juvenile coho salmon behavioural characteristics in Klamath River summer thermal refugia. River Res. Appl. 28, 338–346 (2012).
Miya, M. et al. Use of a filter cartridge for filtration of water samples and extraction of environmental DNA. J. Vis. Exp. JoVE https://doi.org/10.3791/54741 (2016).
Miya, M. et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088 (2015).
O’Donnell, J. L., Kelly, R. P., Lowell, N. C. & Port, J. A. Indexed PCR primers induce template-specific bias in large-scale DNA sequencing studies. PLoS ONE 11, e0148698 (2016).
Porter, T. M. & Hajibabaei, M. MetaWorks: A flexible, scalable bioinformatic pipeline for high-throughput multi-marker biodiversity assessments. PLoS ONE 17, e0274260 (2022).
Alberdi, A., Aizpurua, O., Gilbert, M. T. P. & Bohmann, K. Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods Ecol. Evol. 9, 134–147 (2018).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Carraro, L., Mächler, E., Wüthrich, R. & Altermatt, F. Environmental DNA allows upscaling spatial patterns of biodiversity in freshwater ecosystems. Nat. Commun. 11, 3585 (2020).
Oksanen, J. et al. Vegan: Community Ecology Package. R package version 2.2-1. 2. 1–2. (2018).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2024).
Zuur, A. F., Ieno, E. N., Walker, N., Saveliev, A. A. & Smith, G. M. Mixed Effects Models and Extensions in Ecology with R (Springer, New York, 2009). https://doi.org/10.1007/978-0-387-87458-6.
Pollock, H. S. et al. Long-term monitoring reveals widespread and severe declines of understory birds in a protected Neotropical forest. Proc. Natl. Acad. Sci. 119, e2108731119 (2022).
Lindgren, F., Rue, H. & Lindström, J. An explicit link between Gaussian fields and Gaussian Markov random fields: The stochastic partial differential equation approach: link between Gaussian fields and Gaussian Markov random fields. J. R. Stat. Soc. Ser. B Stat. Methodol. 73, 423–498 (2011).
Zimmerman, D. L. & Ver Hoef, J. M. The Torgegram for fluvial variography: Characterizing spatial dependence on stream networks. J. Comput. Graph. Stat. 26, 253–264 (2017).
Rue, H., Martino, S. & Chopin, N. Approximate Bayesian inference for latent Gaussian models by using integrated nested Laplace approximations. J. R. Stat. Soc. Ser. B Stat. Methodol. 71, 319–392 (2009).
Lindgren, F. & Rue, H. Bayesian spatial modelling with R—INLA. J. Stat. Softw. 63, 1–25 (2015).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
NRC (National Research Council). Endangered and Threatened Fishes in the Klamath River Basin: Causes of Decline and Strategies for Recovery 10838 (National Academies Press, 2004). https://doi.org/10.17226/10838.
Carraro, L., Hartikainen, H., Jokela, J., Bertuzzo, E. & Rinaldo, A. Estimating species distribution and abundance in river networks using environmental DNA. Proc. Natl. Acad. Sci. 115, 11724–11729 (2018).
Winkowski, J. J., Olden, J. D. & Brown, S. Integrating spatial stream network models and environmental DNA to estimate current and future distributions of nonnative Smallmouth Bass. Trans. Am. Fish. Soc. 153, 180–199 (2024).
Ver Hoef, J. M., Peterson, E. E., Clifford, D. & Shah, R. SSN: An R package for spatial statistical modeling on stream networks. J. Stat. Softw. 56, 1–45 (2014).
Struthers, D. P. et al. Statistical stream temperature modelling with SSN and INLA: An introduction for conservation practitioners. Can. J. Fish. Aquat. Sci. 81, 417–432 (2024).
Rodriguez-Ezpeleta, N. et al. Trade-offs between reducing complex terminology and producing accurate interpretations from environmental DNA: Comment on “Environmental DNA: What’s behind the term?” by Pawlowski et al., (2020). Mol. Ecol. 30, 4601–4605 (2021).
Deiner, K. & Altermatt, F. Transport distance of invertebrate environmental DNA in a natural river. PLoS ONE 9, e88786 (2014).
Pess, G. R., Quinn, T. P., Gephard, S. R. & Saunders, R. Re-colonization of Atlantic and Pacific rivers by anadromous fishes: Linkages between life history and the benefits of barrier removal. Rev. Fish Biol. Fish. 24, 881–900 (2014).
Waldman, J. R. & Quinn, T. P. North American diadromous fishes: Drivers of decline and potential for recovery in the Anthropocene. Sci. Adv. 8, eabl5486 (2022).
Munsch, S. H. et al. Dam removal enables diverse juvenile life histories to emerge in threatened salmonids repopulating a heterogeneous landscape. Front. Ecol. Evol. 11, 1188921 (2023).
Fraik, A. K. et al. The impacts of dam construction and removal on the genetics of recovering steelhead (Oncorhynchus mykiss) populations across the Elwha River Watershed. Genes 12, 89 (2021).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016).
Parker, K. A., Hess, J. E., Narum, S. R. & Kinziger, A. P. Evidence for the genetic basis and epistatic interactions underlying ocean- and river-maturing ecotypes of Pacific Lamprey (Entosphenus tridentatus) returning to the Klamath River, California. Mol. Ecol. 28, 15136. https://doi.org/10.1111/mec.15136 (2019).
Zydlewski, J. et al. Seven dam challenges for migratory fish: Insights from the Penobscot River. Front. Ecol. Evol. 11, 1253657 (2023).
Hitt, N. P., Eyler, S. & Wofford, J. E. B. Dam removal increases American eel abundance in distant headwater streams. Trans. Am. Fish. Soc. 141, 1171–1179 (2012).
Leigh, D. M. et al. Best practices for genetic and genomic data archiving. Nat. Ecol. Evol. 8, 1224–1232 (2024).
Nicholson, A. et al. An analysis of metadata reporting in freshwater environmental DNA research calls for the development of best practice guidelines. Environ. DNA 2, 343–349 (2020).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Bergström, A. Improving data archiving practices in ancient genomics. Sci. Data 11, 754 (2024).
ISBER Best Practices. Recommendations for Repositories (International Society for Biological and Environmental Repositories, 2023).
Babel, M. et al. Compared DNA and RNA quality of breast cancer biobanking samples after long-term storage protocols in − 80 °C and liquid nitrogen. Sci. Rep. 10, 14404 (2020).
Acknowledgements
We thank Cheryl Dean for her invaluable support and contributions to molecular analysis for this study. Special thanks to Lauren Frick for assistance with geospatial database management and mapping, and John Lang, Joel Ophoff, Olivia Vosburg, Stephen Staiger, and David Coffman for rugged field navigation expertise and data collection activities. Thanks to the extensive and valued internal review by Leanne Knutson with the Yurok Tribal Fisheries Department. Additionally, we want to acknowledge that the Klamath dam removal was the result of advocacy from the Tribes of the Klamath River Basin, state and federal governments, and the many other organizations. Finally, we extend our gratitude to our reviewers for their insightful feedback and support. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Contributions
DK and DC led the conceptualization of this work, conducted field investigation and data collection, contributed to the original draft, review and editing of the manuscript. KK, GS, and SB led the molecular analysis. DK and KK led the statistical analysis and figure creation. SB, GS, and OO contributed to study concept and design, analysis and interpretation of data, and drafted content. JD and CO contributed to the conceptualization, investigation, original draft, review and editing of the manuscript. All authors approve of this manuscript being submitted when it reaches its final form.
Corresponding authors
Ethics declarations
Competing interests
DK and DC are employed by the restoration contractor for the Klamath River Renewal Project (Lower Klamath Project). Ongoing Lower Klamath Project restoration actions and performance monitoring are expected over the next 6 years and have facilitated the initial establishment of the molecular library. All other authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Keel, D.J., Karpenko, K., Blankenship, S.M. et al. A molecular specimen bank for contemporary and future study captures landscape-scale biodiversity baselines before Klamath River dam removal. Sci Rep 15, 20679 (2025). https://doi.org/10.1038/s41598-025-07042-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-07042-1
