
Abstract
Vitiligo is a complex autoimmune skin disorder characterized by depigmentation and immune dysregulation. To elucidate the role of ferroptosis-related genes (FRGs) in vitiligo, we conducted a comprehensive analysis of gene expression data from the GSE53146 and GSE65127 datasets obtained from the GEO database. We identified 31 differentially expressed FRGs (DE-FRGs), with 21 genes upregulated and 10 downregulated. Functional enrichment analysis revealed that these DE-FRGs are significantly involved in oxidative stress, immune regulation, and vitiligo-associated signaling pathways. Utilizing machine learning approaches, including LASSO and SVM-RFE, we identified four key marker genes (ALOX5, SNCA, SLC1A4, and IL33) with strong diagnostic potential. Immune landscape analysis demonstrated that these marker genes influence immune cell composition, particularly showing correlations with CD8 + T cells and regulatory T cells. Furthermore, drug-gene interaction analysis proposed potential therapeutic targets, while ceRNA network analysis uncovered intricate regulatory relationships involving miRNAs and lncRNAs. Collectively, our findings provide novel insights into the molecular mechanisms underpinning vitiligo and suggest new avenues for diagnostic and therapeutic development.
Similar content being viewed by others

Introduction
Vitiligo is a common chronic autoimmune skin disorder characterized by localized depigmentation, resulting in patches of white skin. The global prevalence of vitiligo is estimated to range from 0.5–2%1. However, recent studies suggested that the actual prevalence may be higher due to underdiagnosis2. Notably, increasing incidence rates among children in the United States and adolescents in Korea indicate that vitiligo is emerging as a significant public health concern3,4. Beyond its physical manifestations, vitiligo profoundly impacts patients’ psychological well-being and quality of life, with disease severity, progression, and duration closely linked to diminished quality of life5. Despite extensive research efforts, the pathogenic mechanisms underlying vitiligo remain incompletely understood. Current evidence suggests that a combination of genetic predisposition, oxidative stress, autoimmunity, inflammation, neurogenic factors, apoptosis, and autophagy contribute to vitiligo pathogenesis6. Central to the disease mechanism is the destruction, death, or dysfunction of melanocytes, highlighting the importance of elucidating the processes that lead to melanocyte loss7. Nevertheless, the precise mechanisms responsible for melanocyte damage remain under debate. Given the unclear pathogenesis and the limited effectiveness of current treatments, there is an urgent need to explore new pathogenic mechanisms in vitiligo to develop effective targeted therapies.
Ferroptosis, a newly recognized type of regulated cell death, is marked by iron-dependent lipid peroxidation, which causes oxidative damage to cell membranes and ultimately leads to cell death8. Recent studies have increasingly explored the potential connection between ferroptosis and vitiligo pathogenesis, reporting significant changes in the expression of ferroptosis markers in the epidermis of vitiligo patients, along with decreased iron levels in peripheral blood9. Additionally, evidence indicates that ferroptosis is extensively involved in autoimmune diseases including vitiligo at the single-cell transcriptome level, further supporting the hypothesis that ferroptosis might contribute to melanocyte destruction in the context of vitiligo10. Furthermore, recent research identified SLC3A2, an RNA-binding protein, as a key regulator of melanocyte ferroptosis in vitiligo, highlighting its critical role in ferroptosis-related pathways and the disease’s pathogenesis11. In vitro experiments have demonstrated that the ferroptosis inducer erastin diminishes the viability of cultured melanocytes, triggers oxidative stress, and leads to the accumulation of iron ions and lipid peroxides9. These findings implicate ferroptosis in melanocyte damage in vitiligo. Additionally, research has demonstrated that RSL3 can induce melanocyte death, mitochondrial dysfunction, reactive oxygen species (ROS) production, and iron accumulation, while baicalein may protect melanocytes from ferroptosis by upregulating GPX4 12. These studies not only underscore the importance of understanding melanocyte ferroptosis in vitiligo but also suggest that targeting this pathway may offer new therapeutic opportunities. Therefore, our study aims to investigate the relationship between ferroptosis, vitiligo, and immune infiltration, focusing on uncovering novel pathogenic mechanisms and identifying potential therapeutic targets.
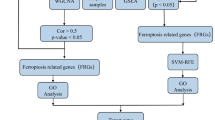
In this study, we aimed to identify key ferroptosis-related genes in vitiligo by integrating differentially expressed genes (DEGs) from the GEO database with ferroptosis-related genes. Through comprehensive bioinformatic analyses, including machine learning approaches and pathway enrichment, we identified potential hub genes associated with vitiligo pathogenesis. Our findings also explored the connection between these hub genes and immune cell infiltration, offering valuable insights into immune dynamics in vitiligo. By identifying potential drug targets and constructing a competing endogenous RNA (ceRNA) network, we aim to provide a foundation for future therapeutic strategies and deeper understanding of vitiligo’s mechanisms.
Results
Identification and analysis of DEGs in vitiligo
To investigate differentially expressed genes (DEGs) in vitiligo lesions, we first acquired raw gene expression data from the GSE53146 and GSE65127 datasets within the GEO database. We then performed comprehensive data preprocessing and cleaning, which included batch effect adjustment and normalization of the expression matrices for both datasets. Visualization using box plots demonstrated consistent trends, with the data closely aligning in nearly straight lines (Fig. 1A, B). Next, we applied a significance threshold of adjusted P < 0.05 and |logFC| > 0.5 to identify DEGs within the merged datasets. These DEGs were visualized in volcano plots (Fig. 1C), and the top 100 DEGs were further highlighted in a heatmap (Fig. 1D).

Identification of DEGs between vitiligo lesions and controls in the combination cohort of GSE53146 and GSE65127 (A) box line blots before homogenization. (B) box line blots after homogenization. (C) volcano map of DEGs. (D) heatmap of the top significantly upregulated or downregulated DEGs.
Identification of DE-FRGs in the combination cohort of GSE53146 and GSE65127
Among the 422 ferroptosis-related genes (FRGs), 31 were identified as differentially expressed between vitiligo patients and healthy controls, with 21 genes up-regulated and 10 down-regulated (Table 1). A clustering heatmap illustrated the expression patterns of these DE-FRGs across the samples (Fig. 2A), while the correlations among these genes were visualized in Fig. 2B.

DE-FRGs Expression Levels in Vitiligo. (A) Violin plots depict the expression patterns of DE-FRGs across various samples. (B) The correlation of these genes.
Functional analyses of DE-FRGs
To explore the functional roles and related signaling pathways of the differentially expressed ferroptosis-related genes (DE-FRGs) in vitiligo, we conducted Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. The GO enrichment analysis revealed significant associations of DE-FRGs with several Biological Processes (BP), including "Response to oxygen levels," "Cellular response to chemical stress," "Response to decreased oxygen levels," "Cellular response to oxidative stress," "Response to oxidative stress," and "Negative regulation of inflammatory response." In the Cellular Components (CC) category, DE-FRGs were enriched in “Nuclear matrix” and "Nuclear periphery," while in Molecular Functions (MF), significant associations were found with "Iron ion binding," "Protein phosphatase binding," and “Phosphatase binding” (Fig. 3A). KEGG pathway analysis indicated that DE-FRGs were significantly enriched in pathways such as "Lipid and atherosclerosis," "Fc epsilon RI signaling pathway," "Sphingolipid signaling pathway," "Efferocytosis," "Hepatitis B," "Diabetic cardiomyopathy," and "MAPK signaling pathway" (Fig. 3B). These findings suggest that DE-FRGs may play critical roles in vitiligo pathogenesis by participating in cellular responses to stress, lipid metabolism, immune regulation, and various kinase-mediated signaling pathways.

Functional analyses for the DE-FRGs revealed significant associations with various biological processes and pathways. (A) GO enrichment analyses indicated that DE-FRGs were significantly related to "Response to oxygen levels," "Cellular response to chemical stress," "Response to decreased oxygen levels," "Cellular response to oxidative stress," "Response to oxidative stress," and "Negative regulation of inflammatory response" in Biological Processes (BP); “Nuclear matrix” and “Nuclear periphery” in Cellular Components (CC); and "Iron ion binding", "protein phosphatase binding", and “phosphatase binding” in Molecular Functions (MF). (B) KEGG pathway enrichment analyses revealed significant relationships with "Lipid and atherosclerosis," "Fc epsilon RI signaling pathway," "Sphingolipid signaling pathway," "Efferocytosis," "Hepatitis B," "Diabetic cardiomyopathy," and "MAPK signaling pathway."
Four DE-FRGs identified as diagnostic genes for vitiligo
To evaluate the potential diagnostic capabilities of DE-FRGs in distinguishing vitiligo patients from healthy controls, we analyzed the merged datasets from GSE53146 and GSE65127 using two machine learning algorithms: LASSO logistic regression and SVM-RFE. The LASSO logistic regression algorithm, with penalty parameter tuning via 10-fold cross-validation, identified 14 vitiligo-related features (Fig. 4A and B). Subsequently, the SVM-RFE algorithm further refined this selection, identifying an optimal combination of 6 DE-FRGs that achieved a maximal accuracy of 0.967 and a minimal RMSE of 0.0333 (Fig. 4C, D). The intersection of marker genes from both LASSO and SVM-RFE models revealed a list of key genes for further analysis (Fig. 4E). A logistic regression model constructed using these genes achieved an AUC of 1.000, demonstrating strong diagnostic capability for vitiligo (Fig. 4F). Additionally, ROC curves generated for the 4 marker genes showed AUC values of 0.738 for ALOX5, 0.849 for SNCA, 0.849 for SLC1A4, and 0.818 for IL33 (Fig. 4G). These findings indicate that the identified DE-FRGs possess significant diagnostic potential for distinguishing vitiligo from healthy samples.

Screening and the diagnostic value of hub genes. (A and B) By LASSO logistic regression algorithm, with penalty parameter tuning conducted by 10-fold cross‐validation, was used to select 14 Vitiligo‐related features. (C and D) SVM‐RFE algorithm to filter the 6 DE‐FRGs to identify the optimal combination of feature genes. Finally, 6 genes (maximal accuracy = 0.967, minimal RMSE = 0.0333) were identified as the optimal feature genes. ( E)The marker genes obtained from the LASSO and SVM‐RFE models. (F) Logistic regression model to identify the AUC of disease samples. (G) ROC curves for the 4 marker genes.
Validation of ferroptosis-related hub genes in clinical skin tissues
To validate the ferroptosis-related hub gene analysis in vitiligo, we measured mRNA expression levels in clinical samples from vitiligo patients relative to healthy controls. The results were largely consistent with our bioinformatics predictions: mRNA levels of SLC1A4 and SNCA were decreased in vitiligo lesions (SLC1A4: 0.6483 ± 0.2674 vs. 1.463 ± 0.1109, mean ± SD, p < 0.05; SNCA: 0.5705 ± 0.2912 vs. 1.764 ± 0.6683, mean ± SD, p < 0.05), while ALOX5 expression was elevated (1.578 ± 0.4818 vs. 0.8038 ± 0.3793, mean ± SD, p < 0.05). However, contrary to our initial bioinformatics insights, IL33 mRNA was significantly higher in vitiligo lesions compared to controls (3.730 ± 0.7253 vs. 1.914 ± 0.5282, mean ± SD, p < 0.05). This divergence highlights the complexity of gene regulation in vitiligo, potentially influenced by patient heterogeneity, disease stage, and treatment history. Such variability is common in autoimmune diseases and may involve mechanisms like epigenetic modifications, post-transcriptional regulation, and signaling pathway crosstalk13,14. s. Larger sample sizes and further studies are required to confirm these findings and clarify the roles of these genes in vitiligo pathogenesis (Fig. 5).

The mRNA expression levels of four ferroptosis-related hub genes in clinical samples from lesions of vitiligo patients and skin tissues from healthy controls. N = 5. The relative expression of mRNA level in the hub genes versus healthy controls is represented on the y-axis. *P < 0.05, **P < 0.01, vs. healthy controls.
Marker genes were closely linked to a variety of vitiligo-related pathways
To determine the functional significance of marker genes in differentiating vitiligo from normal skin, we conducted a comprehensive single-gene Gene Set Enrichment Analysis (GSEA) using KEGG pathways. The top six pathways enriched for each marker gene are illustrated in Fig. 6. This analysis revealed that the marker genes collectively participate in a wide range of biological pathways, highlighting their roles in immune responses, cell signaling, and metabolic processes. The pathways significantly enriched across these genes include immune-related functions such as "Allograft rejection," "Graft-versus-host disease," and "Primary immunodeficiency," as well as pivotal signaling pathways like "Cytokine-cytokine receptor interaction," "Chemokine signaling pathway," "JAK-STAT signaling pathway," and "Hedgehog signaling pathway." Additionally, metabolic processes such as “Steroid biosynthesis” and “Ribosome biogenesis” were prominently featured. This integrative analysis underscores the complex interplay among these genes in modulating immune regulation, signaling pathways, and metabolic functions, suggesting their critical roles in vitiligo pathogenesis.

Single-gene GSEA‐KEGG pathway analysis in four ferroptosis-related hub genes. (A) ALOX5, (B) IL33, (C) SLC1A4, and (D) SNCA.
To further explore the roles of marker genes in vitiligo, we performed Gene Set Variation Analysis (GSVA) to assess differential pathway activation across high- and low-expression groups for each marker gene, as shown in Fig. 7. High expression of ALOX5 was associated with activation of various immune and inflammatory pathways, such as “Leishmania infection” and "RIG-I-like receptor signaling," indicating its crucial role in immune dysregulation in vitiligo. Conversely, low ALOX5 expression correlated with pathways like "TGF-beta signaling" and "Basal cell carcinoma." IL33 exhibited high expression in pathways including "Toll-like receptor signaling" and "Porphyrin and chlorophyll metabolism," reflecting its dual influence on immune and metabolic processes, while low expression was linked to metabolic dysregulation and potential connections to cancer and steroid biosynthesis. Additionally, low SLC1A4 expression was associated with "Pantothenate and CoA biosynthesis" and "Limonene and pinene degradation," pointing to its role in specific metabolic processes. In contrast, high SNCA expression was linked to “GnRH signaling” and "Notch signaling pathways," essential for cellular communication, while low expression influenced metabolic pathways such as "Steroid hormone biosynthesis." These diverse interactions across signaling and metabolic pathways driven by gene expression highlight their profound involvement in vitiligo’s mechanisms, offering deeper insights and potential targets for therapeutic intervention.

High- and low‐expression groups based on the expression levels of each marker gene combined with GSVA in ALOX5 (A), IL33 (B), SLC1A4 (C), and SNCA (D).
Immune landscape analysis
Building on previous findings that linked marker genes to the immune response and underscored their crucial role in the immune microenvironment of vitiligo, we employed the CIBERSORT algorithm to investigate differences in immune cell composition between vitiligo patients and healthy controls. Our analysis revealed significant variations in CD8 T cells (p = 0.008) (Fig. 8A). Subsequent Pearson correlation analysis, presented in Fig. 8B, showed that SNCA was strongly positively correlated with "mast cells resting" (r = 0.354, p < 0.05) and negatively correlated with “plasma cells” (r = -0.284, p < 0.05). Additionally, IL33 and SLC1A4 were negatively correlated with "T cells CD4 naive" (r = -0.312, p < 0.05) and "T cells regulatory (Treg)" (r = -0.276, p < 0.05), respectively. These findings demonstrate the significant influence of these marker genes on the immune microenvironment, reinforcing their involvement in vitiligo pathogenesis.

Immune Landscape Analysis. (A) Utilizing the CIBERSORT algorithm, we identified notable differences in the immune cell composition between vitiligo patients and normal samples, with significant disparities observed in CD8 T cells (p = 0.008).(B) Pearson correlation analysis highlighted significant correlations between marker genes and specific immune cells: SNCA positively correlated with "mast cells resting" and negatively with “plasma cells”; IL33 and SLC1A4 showed negative correlations with "T cells CD4 naive" and "T cells regulatory (Treg)" respectively (*p < 0.05).
Prediction of marker gene-targeted drugs
We explored potential pharmacological interventions targeting the identified marker genes using the DGIdb database and analyzed drug-gene interaction relationships with default settings, as detailed in Table S5. Visualization of these interactions via Cytoscape software is shown in Fig. 9. Our analysis identified a significant number of drugs targeting ALOX5 (25 drugs) and SNCA (28 drugs). These findings suggest that a diverse array of pharmaceutical agents, including inhibitors, could specifically target and modulate the expression of these marker genes. This supports the potential application of these drugs in developing therapeutic strategies for disorders associated with dysregulated ALOX5 and SNCA expression.

Prediction of marker gene-targeted drugs. The drugs may target marker genes through the DGIdb database and the interaction relationship between the two.
A ceRNA network based on marker genes
Using the starBase and miRanda databases, we constructed a comprehensive competing endogenous RNA (ceRNA) network for four vitiligo-related marker genes, as detailed in Fig. 10. This network comprises 264 nodes, including 4 marker genes, 140 miRNAs, and 157 lncRNAs, and features numerous interaction edges that illustrate complex regulatory dynamics. Notably, SLC1A4 is influenced by 46 miRNAs, with some competing with various lncRNAs to regulate its expression. Similarly, SNCA is affected by 53 miRNAs through lncRNA-mediated mechanisms, IL33 is targeted by 36 miRNAs, and ALOX5 interacts with 5 miRNAs.

A ceRNA networks based on marker genes.
Further exploration of the network dynamics reveals that MUC19 modulates ALOX5 expression by binding with hsa-miR-29a-5p and hsa-miR-145-5p, while also influencing IL33 via hsa-miR-382-5p. LINC00969 regulates ALOX5 by associating with hsa-miR-193a-3p and impacts SLC1A4 alongside other lncRNAs through hsa-miR-484. Additionally, complexes involving SLC8A1-AS1, CTA-392E5.1, and RP11-96K19.4 coordinate the regulation of SNCA and IL33 through hsa-miR-335-3p, while RP11-673P17.2, CTB-171A8.1, and RP5-902P8.10 target SNCA and SLC1A4 by binding to hsa-miR-767-3p. This detailed map of lncRNA-miRNA interactions provides a deeper understanding of the regulatory mechanisms that may influence vitiligo pathogenesis, opening novel avenues for therapeutic intervention. Further details of these interactions and the network configuration are described in Table S6.
Discussion
In the context of vitiligo pathogenesis, ferroptosis, a programmed form of cell death, emerges as a potential contributor to melanocyte demise. Ferroptosis involves three key processes: metabolic mechanisms, regulation of reactive oxygen species (ROS), and control of intracellular iron levels15. Previous studies have highlighted the roles of ROS, oxidative stress, and autophagy in the pathogenesis of vitiligo16. Traditionally, melanocyte death has been primarily associated with apoptosis; however, our hypothesis suggests a possible involvement of ferroptosis in this process. Research indicates a direct link between oxidative stress and ferroptosis, potentially mediated through the NRF2-ARE pathway17. Additionally, ROS-mediated autophagy, regulated by ferritin and the transferrin receptor, modulates intracellular iron levels and ferroptosis17.
The growing body of evidence increasingly supports the involvement of ferroptosis in vitiligo’s pathogenic mechanisms. Studies have shown significant alterations in the expression of ferroptosis markers within the epidermis of vitiligo patients9. Moreover, ferroptosis has been identified as a factor contributing to the loss of melanocytes during vitiligo development10. It has also been observed that interferon-gamma (IFN-γ) can regulate cell death pathways, particularly through oxidative stress-induced ferroptosis18. However, the role of ferroptosis in other cell types involved in vitiligo, such as keratinocytes and fibroblasts, remains largely unexplored. Furthermore, there has been little documentation of bioinformatic analyses focusing on ferroptosis-related genes in vitiligo. Understanding the role of ferroptosis in melanocytes could offer valuable insights into the broader pathogenesis of vitiligo. Conducting bioinformatic analyses to elucidate the molecular pathways and genetic factors associated with ferroptosis in vitiligo may reveal novel therapeutic targets and diagnostic biomarkers for the disease.
In this study, utilizing LASSO and SVM-RFE algorithms, we identified four key genes associated with ferroptosis in vitiligo: ALOX5, IL33, SNCA, and SLC1A4. These genes exhibited relatively high AUC values in the combined dataset (ALOX5, AUC = 0.738; SNCA, AUC = 0.849; SLC1A4, AUC = 0.849; IL33, AUC = 0.818), indicating their potential as specific biomarkers for diagnosing vitiligo. Q-PCR validation experiments revealed elevated expression of ALOX5 and decreased expression of SNCA and SLC1A4 in vitiligo patients compared to healthy controls, consistent with our bioinformatic analysis. However, contrary to our expectations, IL33 expression levels were found to be elevated in vitiligo patients. We hypothesize that this discrepancy may reflect a dual role of IL33 in the pathogenesis of vitiligo. Previous studies have reported increased IL-33 expression in lesional skin of vitiligo patients, along with elevated serum levels. On one hand, IL-33, acting as an alarmin, could potentially induce melanocyte death by modulating cytokine expression in the cellular microenvironment, thus contributing to vitiligo pathogenesis19. On the other hand, in other inflammatory diseases, IL-33 has been shown to provide acute protection by acting as an alarmin while also mediating long-term immunosuppression and immune dysfunction via Tregs, highlighting its tissue-specific roles in immune regulation20. This duality might also exist in vitiligo, warranting further studies to clarify the specific conditions under which IL-33 functions as a pro-inflammatory factor versus a regulatory modulator, to better understand its precise role in vitiligo pathogenesis. Furthermore, the positive correlation between serum IL-33 levels and the extent and activity of vitiligo, consistent with our validation results, underscores the significance of IL33 in vitiligo development21.
Several factors may contribute to the observed discrepancy between our validation results and the combined dataset regarding IL33 expression. Firstly, the heterogeneity of vitiligo patients, including variations in disease stage, duration, and treatment history, may influence IL33 expression levels. Secondly, the complexity of IL33 signaling pathways and its interactions with other molecules in the skin microenvironment could result in diverse expression patterns among different individuals. Additionally, technical variations in qPCR assays, sample quantity, and processing methods may impact the accuracy of gene expression measurements.
SNCA encodes α-synuclein (aSyn), a protein implicated in neuronal pigmentation. Previous in vitro studies have shown that downregulation of aSyn leads to a reduction in melanosome release and transfer to keratinocytes, suggesting its involvement in pigment deposition in vitiligo22. Additionally, in melanoma, SNCA has been found to promote tumor cell proliferation and support tumor cell survival23,24, indicating a potential role in regulating melanoma cell growth. Notably, studies have demonstrated that knocking out SNCA in melanoma cells results in decreased expression of transferrin receptor 1 (TfR1), increased ferritin levels, and elevated reactive oxygen species, accompanied by reduced proliferation rates compared to control cells25. This suggests that SNCA may be involved in regulating iron-mediated cell death processes, such as ferroptosis, in melanoma cells. Given these findings, SNCA holds significant research potential for understanding melanocyte metabolism and iron-mediated cell death pathways in vitiligo. Future studies are warranted to confirm its role in modulating melanocyte function and iron-mediated cell death.
The SLC1A4 gene encodes a sodium-dependent neutral amino acid transporter that facilitates the transport of alanine, serine, cysteine, and threonine26. SLC1A4 has been implicated as a gene associated with ferroptosis in various tumor types27. Additionally, studies have identified ALOX5 as a reliable predictor of overall survival in melanoma28, with ALOX5 deficiency conferring resistance to autophagy and ferroptosis by inhibiting the AMPK/mTOR pathway in melanoma cells29. In our investigation, we observed elevated expression of ALOX5 in vitiligo patients compared to healthy controls, suggesting a potential role for ALOX5 in the pathogenesis of vitiligo, potentially through the induction of ferroptosis in melanocytes.
Vitiligo, characterized by an autoimmune response, involves dysregulation of immune cells. Previous studies have highlighted the pivotal role of CD8 + T cells in vitiligo pathogenesis, with autoreactive CD8 + T cells driving immune responses and inducing melanocyte apoptosis through IFN-γ secretion30. Elevated levels of CD8 + T cells in peripheral blood and their correlation with disease severity further underscore their significance in vitiligo progression31,32. Our study corroborates these findings, revealing notable differences in immune cell composition between vitiligo patients and healthy controls, particularly in CD8 + T cells, emphasizing their central role in vitiligo pathogenesis. Additionally, our analysis uncovered significant correlations between marker genes and specific immune cell populations. Notably, SNCA exhibited positive correlations with resting mast cells and negative correlations with plasma cells. IL33 and SLC1A4 displayed negative correlations with naïve CD4 + T cells and regulatory T cells (Tregs), respectively. These immune cell alterations observed in vitiligo lesions might suggest an overactive immune response contributing to melanocyte destruction. However, further studies are required to validate these associations and clarify the specific mechanisms through which immune cell dysregulation impacts melanocyte survival.
In this study, we analyzed marker genes for gene-targeted drugs and the ceRNA network. Among the 25 ALOX5-targeted drugs identified, Zileuton emerged as an FDA-approved inhibitor of ALOX5. Given the pivotal role of 5-lipoxygenase (encoded by ALOX5) in immune regulation, Zileuton modulates tumor-associated macrophage M2 polarization via the JAK/STAT pathway, thereby promoting invasion and metastasis in pancreatic cancer, suggesting its potential value in tumor therapy33. Additionally, Zileuton demonstrates alleviation of depressive-like behaviors and neuroinflammation34. As a novel anti-inflammatory compound, Zileuton may potentially replace systemic antibiotics in the future treatment of severe acne, indicating its therapeutic potential in skin inflammatory diseases35. Furthermore, Sulfasalazine, another inhibitor of ALOX5, has demonstrated its ability to suppress the release of pro-inflammatory cytokines such as IL-1, IL-6, IL-12, and TNF36. Notably, in patients with active vitiligo, there is a significant elevation in TNF-α and IL-6 levels37. Sulfasalazine also shows potential therapeutic value in treating recalcitrant cases of alopecia areata38, an autoimmune disease that shares common pathogenic mechanisms with vitiligo39. Consequently, Sulfasalazine emerges as a promising candidate for vitiligo treatment, underscoring the need for further fundamental research to validate its therapeutic potential.
Non-coding RNAs (ncRNAs) have also emerged as pivotal players in the development of vitiligo. Specifically, miR-211 has been identified as a regulator of mitochondrial energy metabolism in vitiligo, while melanogenesis-associated miRNAs such as miR-423, miR-106a, and miR-182 are implicated in the pathogenesis of vitiligo and hold promise as potential biomarkers for the condition40,41. Additionally, miR-9 has been found to regulate melanocyte adhesion and migration during vitiligo repigmentation induced by UVB treatment42. Recent research has illuminated the regulatory roles of long non-coding RNAs (lncRNAs) in vitiligo pathogenesis. Notably, the lncRNA MALAT1 has been identified as a significant contributor to vitiligo pathophysiology. By inhibiting miR-211, MALAT1 facilitates the upregulation of sirtuin 1 expression, thereby protecting the vitiligo epidermis against DNA damage induced by UV radiation43. Moreover, elevated expression of lncRNA LOC100506314 in CD4 + T cells isolated from vitiligo patients has been positively correlated with disease severity44.
These findings collectively underscore the emerging role of microRNAs and lncRNAs in vitiligo pathogenesis, highlighting their potential clinical utility as biomarkers and therapeutic targets for vitiligo patients. However, the precise roles of the gene-targeted drugs and non-coding RNAs predicted in our study remain ambiguous, necessitating further investigation into their specific pathways. Therefore, prospective studies are warranted to delve deeper into these selected drugs and non-coding RNAs, unveiling their mechanisms of action and therapeutic potential.
This study also has several limitations that should be acknowledged. First, the datasets used in this analysis are relatively small, which may limit the generalizability of the findings. Second, while we performed qPCR validation, further functional experiments are needed to confirm the biological relevance of the identified hub genes. Additionally, patient heterogeneity, including differences in disease stage, duration, and treatment history, may have influenced the observed gene expression patterns. Future research with larger datasets, diverse patient populations, and mechanistic studies is necessary to validate and extend our findings.
Methods and materials
Data source
In this study, gene expression data for active vitiligo and healthy controls were obtained from the Gene Expression Omnibus (GEO) database. The dataset GSE53146 included 5 vitiligo patients and 5 healthy individuals, while GSE65127 consisted of 10 vitiligo patients and 10 healthy controls. The matrix data, along with the corresponding chip platform annotation files for GSE65127 and GSE53146, were downloaded for analysis. Each expression matrix was subsequently log2 transformed using R language, and probe IDs were converted to gene symbols. Normalization of each chip dataset was performed using the ‘limma’ package in R. To address batch effects arising from the use of different platforms for the vitiligo chip datasets, we employed Strawberry Perl software version 5.30.0.1 (http://strawberryperl.com/) for batch merging and the ‘limma’ and ‘sva’ packages in R for batch effect correction. The resulting merged datasets were consolidated into a new dataset, which served as the training set for this study. We acknowledge that the datasets used in this analysis are relatively small. This limitation should be considered.
Additionally, the ferroptosis-related genes (FRGs; n = 844) used in this study were sourced from FerrDb (http://www.zhounan.org/ferrdb/current/), with the detailed gene list provided in Table S1.
The Drug Gene Interaction Database (DGIdb) was utilized to predict drugs targeting the identified marker genes, while the structural information for these targeted drugs was obtained from the DrugBank database.
Collection of skin samples
This study received ethical approval from the Ethics Committee of Suining Central Hospital. All methods were performed in accordance with relevant ethical guidelines and regulations. Skin tissue samples were obtained from the lesions of 5 active vitiligo patients and 5 healthy controls, all recruited through the Dermatology Department of Suining Central Hospital. The 5 vitiligo patients included in the RT-PCR study were selected to match the clinical characteristics of the vitiligo patients in the differential gene expression (DGE) studies Written informed consent was obtained from all participants before sample collection. The collected skin tissues from both vitiligo patients and healthy controls were promptly preserved in a -80 °C freezer for subsequent analysis.
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted from skin tissue samples using TRIzol reagent (AG21101, AG). cDNA synthesis was performed using the HiScript III RT SuperMix for qPCR kit with gDNA removal (RR0701, AboRo, Shenzhen, China). Real-time PCR was conducted using the SYBR qPCR kit (RS0601, AboRo, Shenzhen, China). The primer sequences used in this study are listed in Supplementary Table S2. The mRNA expression levels of ferroptosis-related hub genes were analyzed using the LightCycler 480 system (Roche, Basel, Switzerland). The relative transcriptional levels of these genes, normalized to GAPDH (Tsingke, Beijing, China), were calculated using the 2 − ΔΔCt method.
For the RT-PCR analysis, triplicate technical replicates were performed for each gene. The quantitative data are presented as mean ± standard deviation (SD). Statistical analysis and graphing were performed using GraphPad Prism 10 software. Non-paired t-tests were used to compare differences between groups, and p-values were considered statistically significant at p < 0.05.
Differential expression analysis
The expression profiles from GSE53146 and GSE65127 were merged into a consolidated microarray dataset. To address batch effects, we utilized the R packages “limma” and "sva," while “preprocessCore” was employed for data homogenization. Subsequently, differentially expressed genes (DEGs) were identified in skin tissue samples from vitiligo lesions and controls using the “limma” package, with DEGs defined as those having adjusted P-values < 0.05 and |logFC| > 0.5. Heatmaps were generated using the “heatmap” and “ggplot2” packages in R.
We further extracted expression data for 422 ferroptosis-related genes (FRGs) from normal and vitiligo samples within the merged GSE53146 and GSE65127 dataset (only 422 FRGs expressed in the merged dataset). Differential expression analysis was performed using Student’s t-test in R, and genes with a significance level of p < 0.05 were considered noteworthy (see Table S3).
Functional analysis of DEGs
To investigate the role of DEGs in vitiligo and ferroptosis, GO and KEGG analyses were performed using the “clusterProfiler” program. A P-value of less than 0.05 was considered statistically significant.
Identification and ROC curves of hub genes
The LASSO (Least Absolute Shrinkage and Selection Operator) algorithm, implemented via the “glmnet” package, was used to streamline the dataset by focusing on differentially expressed ferroptosis-related genes (DE-FRGs) between vitiligo patients and healthy controls, identifying potential gene biomarkers for vitiligo. Simultaneously, a support vector machine-recursive feature elimination (SVM-RFE) model was developed using the “SVM” package. The efficacy of the SVM-RFE model was compared with the LASSO model by evaluating their average misclassification rates through 10-fold cross-validation. Common genes identified by both models were considered optimal biomarkers. The diagnostic capabilities of these biomarkers were validated using receiver operating characteristic (ROC) curves, with assessments of area under the curve (AUC), accuracy, sensitivity, and specificity. Additionally, a logistic regression model incorporating four marker genes was constructed to predict sample types within the combined GSE65127 and GSE53146 dataset, with its diagnostic performance assessed through ROC curve analysis.
Single-gene gene set enrichment analysis (GSEA) enrichment analysis
The analysis was conducted using the GSEA (v4.1.0) package in R. To explore pathways associated with the four marker genes, we calculated their correlations with all other genes in the merged GSE53146 and GSE65127 dataset. These genes were then ranked in descending order based on their correlation values, forming the test gene set. The KEGG signaling pathway set was used as a predefined set to evaluate its enrichment within this ranked gene set.
Single-gene gene set variation analysis (GSVA) enrichment analysis
The analysis was performed using the GSVA (Gene Set Variation Analysis) package (v1.38.0) in R. The KEGG pathway set was used as the reference gene set for conducting GSVA on each marker gene. Concurrently, the limma package was applied to evaluate differences in GSVA scores between high- and low-expression groups of the marker genes, with |t| > 2 and p < 0.05 as the significance criteria. A positive t-value indicated pathway activation in the high-expression group, while a negative t-value suggested activation in the low-expression group.
Immune infiltration analysis
The analysis was performed using CIBERSORT, a tool designed to estimate the cellular composition of complex tissues based on gene expression profiles. Using CIBERSORT, we assessed the proportions of 22 different immune cell types in each tissue sample from the merged GSE53146 and GSE65127 dataset. The proportions of all immune cell types in each sample were normalized to sum to 1 (Table S4).
Construction of ceRNA network
The starBase database was utilized to predict mRNA-miRNA interaction pairs for seven marker genes. RNA sequences for four of these marker genes were obtained from the National Center for Biotechnology Information (NCBI), and human miRNA sequences were sourced from miRBase. To enhance the stringency of mRNA-miRNA interaction predictions, the binding score threshold in miranda software was increased from the default 140 to 170. Subsequent searches in starBase enabled the identification and screening of miRNA-lncRNA interactions, ultimately establishing the ceRNA network comprising mRNA-miRNA-lncRNA relationships.
Statistical analysis
Comparative analysis between the two groups was performed using Student’s t-test. The associations among 31 differentially expressed ferroptosis-related genes (DE-FRGs) were examined using Pearson correlation analysis. Venn diagrams were generated with the Jvenn package, and the ceRNA network was visualized using Cytoscape. A p-value of less than 0.05 was considered statistically significant. All statistical analyses were conducted in R.
Data availability
The datasets used in this study are available from the corresponding author upon reasonable request.
References
Bergqvist, C., Ezzedine, K. & Vitiligo Rev. Dermatology 236, 571–592 (2020).
Gandhi, K. et al. Prevalence of Vitiligo among adults in the United States. JAMA Dermatol. 158, 43–50 (2022).
Patel, R. et al. Prevalence of Vitiligo among children and adolescents in the United States. Dermatology 239, 227–234 (2023).
Kang, H. & Lee, S. Prevalence and incidence of vitiligo and associated comorbidities: a nationwide population-based study in Korea. Clin. Exp. Dermatol. 48, 484–489 (2023).
Bibeau, K. et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J. Eur. Acad. Dermatol. Venereol. 36, 1831–1844 (2022).
Iwanowski, T., Kołkowski, K., Nowicki, R. J. & Sokołowska-Wojdyło, M. Etiopathogenesis and emerging methods for treatment of Vitiligo. Int. J. Mol. Sci. 24, 9749 (2023).
Liu, L. Y. et al. The role of regulatory cell Death in Vitiligo. DNA Cell. Biol. 43, 61–73 (2024).
Dixon, S. J. & Olzmann, J. A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell. Biol. 25 (6), 424–442 (2024).
Wu, X. et al. Altered expression of ferroptosis markers and iron metabolism reveals a potential role of ferroptosis in vitiligo. Pigment Cell. Melanoma Res. 35, 328–341 (2022).
Zhang, D. et al. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J. Transl Med. 20, 363 (2022).
Zhang, J. et al. Identification and validation of RNA-binding protein SLC3A2 regulates melanocyte ferroptosis in vitiligo by integrated analysis of single-cell and bulk RNA-sequencing. BMC Genom. 25, 236 (2024).
Yang, M. et al. Baicalein inhibits RLS3-induced ferroptosis in melanocytes. Biochem. Biophys. Res. Commun. 561, 65–72 (2021).
Post, N. F. et al. Trained immunity in the pathogenesis of vitiligo. Pigment Cell. Melanoma Res. 36, 348–354 (2023).
Gibson, F. et al. Epigenetic dysregulation in autoimmune and inflammatory skin diseases. Clin. Rev. Allergy Immunol. 63, 447–471 (2022).
Stockwell, B. R. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Chen, J., Li, S. & Li, C. Mechanisms of melanocyte death in vitiligo. Med. Res. Rev. 41, 1138–1166 (2021).
Chen, G. H. et al. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol. Med. 180, 95–107 (2022).
Ng, C. Y. et al. Targeting the elevated IFN-γ in vitiligo patients by human anti- IFN-γ monoclonal antibody hampers direct cytotoxicity in melanocyte. J. Dermatol. Sci. 110, 78–88 (2023).
Li, P., Ma, H., Han, D. & Mou, K. Interleukin-33 affects cytokine production by keratinocytes in vitiligo. Clin. Exp. Dermatol. 40, 163–170 (2015).
Arpaia, N. et al. A distinct function of regulatory T Cells in tissue protection. Cell 162, 1078–1089 (2015).
Vaccaro, M. et al. IL-33 circulating serum levels are increased in patients with non-segmental generalized vitiligo. Arch. Dermatol. Res. 308, 527–530 (2016).
Rachinger, N. et al. Alpha-Synuclein and Its Role in Melanocytes. Cells. 11, 2087 (2022).
Niederberger, E. et al. Distinct molecular mechanisms contribute to the reduction of melanoma growth and tumor pain after systemic and local depletion of alpha-synuclein in mice. FASEB J. 37, e23287 (2023).
Gajendran, N., Rajasekaran, S. & Witt, S. N. Knocking out alpha-synuclein in melanoma cells downregulates L1CAM and decreases motility. Sci. Rep. 13, 9243 (2023).
Shekoohi, S. et al. Knocking out alpha-synuclein in melanoma cells dysregulates cellular iron metabolism and suppresses tumor growth. Sci. Rep. 11, 5267 (2021).
Scalise, M. et al. ASCT1 and ASCT2: brother and sister? SLAS Discov. 26, 1148–1163 (2021).
Zhang, L. et al. Ferroptosis regulator NOS2 is closely associated with the prognosis and cell malignant behaviors of hepatoblastoma: a bioinformatic and in vitro study. Front. Oncol. 13, 1228199 (2023).
Zeng, N. et al. Construction of a Ferroptosis-related gene signature for predicting survival and immune microenvironment in melanoma patients. Int. J. Gen. Med. 14, 6423–6438 (2021).
Wang, M. et al. ALOX5 promotes autophagy-dependent ferroptosis by activating the AMPK/mTOR pathway in melanoma. Biochem. Pharmacol. 212, 115554 (2023).
Harris, J. E. et al. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8⁺ T-cell accumulation in the skin. J. Invest. Dermatol. 132, 1869–1876 (2012).
Bergqvist, C., Ezzedine, K. & Vitiligo A focus on pathogenesis and its therapeutic implications. J. Dermatol. 48, 252–270 (2021).
Strassner, J. P., Rashighi, M., Ahmed Refat, M., Richmond, J. M. & Harris, J. E. Suction blistering the lesional skin of vitiligo patients reveals useful biomarkers of disease activity. J. Am. Acad. Dermatol. 76, 847–855 (2017).
Hu, W. M. et al. The ALOX5 inhibitor Zileuton regulates tumor-associated macrophage M2 polarization by JAK/STAT and inhibits pancreatic cancer invasion and metastasis. Int. Immunopharmacol. 121, 110505 (2023).
Liu, C. H. et al. Zileuton ameliorates depressive-like behaviors, hippocampal neuroinflammation, apoptosis and synapse dysfunction in mice exposed to chronic mild stress. Int. Immunopharmacol. 78, 105947 (2020).
Zouboulis, C. C. & Bettoli, V. Management of severe acne. Br. J. Dermatol. 172 (Suppl 1), 27–36 (2015).
Mushtaq, S. & Sarkar, R. Sulfasalazine in dermatology: a lesser explored drug with broad therapeutic potential. Int. J. Womens Dermatol. 6, 191–198 (2020).
Karagün, E. & Baysak, S. Levels of TNF-α, IL-6, IL-17, IL-37 cytokines in patients with active vitiligo. Aging Male. 23, 1487–1492 (2020).
Aghaei, S. An uncontrolled, open label study of sulfasalazine in severe Alopecia Areata. Indian J. Dermatol. Venereol. Leprol. 74, 611–613 (2008).
Yamaguchi, H. L., Yamaguchi, Y. & Peeva, E. Pathogenesis of Alopecia Areata and Vitiligo: commonalities and differences. Int. J. Mol. Sci. 25, 4409 (2024).
Spiegelman, V. S., Elcheva, I. A. & Metabo-miR miR-211 regulates mitochondrial energy metabolism in Vitiligo. J. Invest. Dermatol. 137, 1828–1830 (2017).
Abdallah, H. Y. et al. Investigating melanogenesis-related microRNAs as disease biomarkers in vitiligo. Sci. Rep. 12, 13526 (2022).
Su, M. et al. miR-9 regulates melanocytes adhesion and migration during vitiligo repigmentation induced by UVB treatment. Exp. Cell. Res. 384, 111615 (2019).
Brahmbhatt, H. D. et al. The long noncoding RNA MALAT1 suppresses miR-211 to confer protection from ultraviolet-mediated DNA damage in vitiligo epidermis by upregulating sirtuin 1. Br. J. Dermatol. 184, 1132–1142 (2021).
Lai, N. S., Yu, H. C., Huang, H. B., Huang Tseng, H. Y. & Lu, M. C. Increased Expression of Long Noncoding RNA LOC100506314 in T cells from Patients with Nonsegmental Vitiligo and Its Contribution to Vitiligo Pathogenesis. Mediators Inflamm. 2440377 (2023). (2023).
Acknowledgements
We extend our heartfelt gratitude to the patients who generously provided their consent for the use of their clinical data in this study. We also thank the Suining Central Hospital Basic Laboratory for providing the experimental platform.
Funding
This work was supported by the Sichuan Medical Youth Innovation Research Project, Grant/Award Number: Q23014; Suining Health Science and Technology Project, Grant/Award Number: 24CJDFB07.
Author information
Authors and Affiliations
Contributions
Linli Liu and Chunshui Yu designed the project. Linli Liu conducted the genetic and bioinformatic analyses. Li Guan performed the laboratory experiments. Lingli Deng collected the clinical data. Yuan Hu and Qianying Li made the data graph. Linli Liu, Lingli Deng, and Li Guan contributed equally to this work. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of Suining Central Hospital and written consents from the patients were obtained.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, L., Deng, L., Guan, L. et al. Bioinformatic analysis of ferroptosis related biomarkers and potential therapeutic targets in vitiligo. Sci Rep 15, 2035 (2025). https://doi.org/10.1038/s41598-025-86061-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-86061-4
