
Abstract
Non-small cell lung cancer (NSCLC) remains a leading cause of cancer-related mortality, with immunotherapy proving effective only in a subset of patients, highlighting the urgent need for improved biomarkers and therapeutic targets. This study explored regulatory T cell (Treg) metabolism-related genes in NSCLC and established a seven-gene prognostic signature (TIMP1, BIRC3, G0S2, PRKCB, PDE4B, CD52, and ACP5) through multi-omics analysis. The model stratified patients into high- and low-risk groups exhibiting distinct immune profiles and drug sensitivities. Clinical validation confirmed that elevated BIRC3 and G0S2 expression correlated with poorer prognosis, while functional assays demonstrated that G0S2 inhibition suppressed tumour progression and reduced Treg infiltration in vivo. These findings position G0S2 as a promising biomarker for immunotherapy response and a potential therapeutic target, providing insights into Treg-mediated immune regulation and advancing personalized NSCLC treatment strategies.

Similar content being viewed by others
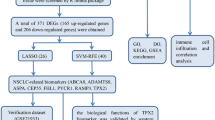

Introduction
Lung cancer is the leading cause of cancer-related mortality, accounting for approximately 21% of all such deaths; notably, NSCLC is the main cause of mortality among lung cancer patients and accounts for 85% to 90% of lung cancer cases1. Despite advancements in diagnostics, many patients are diagnosed at an advanced stage, which limits surgical options. Recent developments in targeted therapies and immunotherapies, particularly tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs), have significantly improved the survival rates of patients in advanced stages2,3. However, these treatments are effective only in specific patient groups, and their efficacy is greatly limited by factors such as drug resistance and tumour immune escape4. This underscores the urgent need for continued research into identifying potential biomarkers and developing novel therapeutic targets to gain a deeper understanding of NSCLC.
Tregs, a subset of immunosuppressive CD4+ T cells, play a critical role in tumour immune regulation. Tregs suppress the function of CD8+ and CD4+ effector T cells by secreting inhibitory cytokines such as IL-10 and TGF-β, competitively consuming IL-2, and inducing adenosine production, thereby attenuating anti-tumour immune responses5,6,7. Given their immunosuppressive function, Tregs are increasingly recognized as a promising immunotherapeutic target in NSCLC. Currently, multiple strategies aimed at disrupting Treg-mediated immunosuppression are under active investigation. These approaches include the use of immune checkpoint inhibitors such as anti-PD-1, anti-PD-L1 and anti-CTLA-4 antibodies to partially modulate Treg activity; selective depletion of intratumoural Tregs through anti-CD25 antibodies or TNFRSF family agonists such as OX40 and GITR; application of IL-2 variants to preferentially expand effector T cells while avoiding Treg activation; and inhibition of the adenosine signaling pathway to alleviate immunosuppression within the tumour microenvironment8,9,10,11,12. Although these strategies have demonstrated certain anti-tumour potential in preclinical and clinical studies, several challenges remain. First, the lack of exclusive surface markers makes it difficult to distinguish Tregs from activated effector T cells, which compromises targeting specificity and may impair beneficial immune responses. Second, excessive depletion of Tregs may disrupt immune tolerance and lead to severe immune-related adverse events (irAEs). Moreover, Tregs within the tumour microenvironment (TME) are not a homogeneous population; rather, they exhibit considerable functional heterogeneity and phenotypic plasticity across different tumour types, disease stages, and therapeutic contexts13. Therefore, precise identification and selective targeting of functionally distinct Treg subsets represent a critical direction for improving immunotherapy outcomes in NSCLC.
To translate these mechanistic insights into clinically relevant observations, we examined the relationship between immunotherapy efficacy and Treg populations in NSCLC patients receiving immune checkpoint blockade. Patients were categorized into immunotherapy-sensitive and -insensitive groups on the basis of their clinical response. Peripheral blood samples were evaluated to assess the distribution of various immune cell populations, including Tregs, CD3+ T cells, CD4+ T cells, CD8+ T cells, B cells, and NK cells, to explore their potential associations with immunotherapy outcomes. However, our study revealed that the proportions of Tregs in peripheral blood do not effectively predict patient responses to immunotherapy, suggesting that the immunosuppressive activity of circulating Tregs in the tumour microenvironment may not be adequately reflected by this parameter. This finding underscores the importance of exploring Treg cell subtypes in greater depth.
In the tumour microenvironment, metabolic changes are crucial for regulating Treg cell function, which influences tumour development and progression14. Research indicates that metabolites in the tumour microenvironment (such as lactate) significantly affect Treg cell function and the metabolic state15. Lactate not only provides energy for Tregs but also enhances their immunosuppressive capabilities, promoting their survival and functional maintenance in immunosuppressive environments. Additionally, Tregs can regulate Foxp3-dependent RNA splicing by taking up lactate, which in turn affects the expression of CTLA-4 and further modulates Treg cell activity16. In addition to lactate metabolism, lipid metabolism is also closely related to Treg cell function and tumour immune evasion17. Mitochondrial changes in Tregs may be linked to their enhanced immunosuppressive characteristics within the tumour microenvironment. Although the relationship between Tregs and metabolism is becoming clearer, their specific roles in NSCLC still require further investigation. On the basis of these findings, we hypothesize that focusing on the metabolic characteristics of Tregs could reveal new biomarkers and ultimately improve the prognosis of NSCLC patients.
In this study, we aimed to develop a prognostic model and identify potential therapeutic targets for NSCLC to improve treatment and patient management. Most existing studies rely solely on bulk RNA sequencing without considering cellular heterogeneity18. To address this, we integrated single-cell RNA sequencing (scRNA-seq) data with bulk RNA sequencing data and performed other functional experiments for verification. Overall, we constructed and validated a Treg cell metabolism-related gene prognostic model to enhance the understanding of tumour heterogeneity, facilitate prognostic stratification and improve personalized treatment for NSCLC patients.
Results
Peripheral blood lymphocyte subset distribution in NSCLC patients
These patients were then categorized into two groups according to their initial treatment response: treatment-response group (partial response, PR, n = 31) and treatment-non-response group (disease progression, PD, and stable disease, SD, n = 29) (Fig. 1A). We assessed the proportions of Tregs, CD3+ T cells, CD4+ T cells, CD8+ T cells, B cells, and NK cells. The results revealed that the number of Tregs in the treatment-response group did not significantly differ before and after treatment (P = 0.1255), similarly, the proportion of Tregs in the treatment-response group did not significantly differ before and after treatment (P = 0.2228) (Fig. 1B). In the treatment-response group, the proportions of CD4+ T cells (P = 0.0285) and CD8+ T cells (P = 0.0030) were significantly different before and after treatment, while those in non-response group were not significantly different (Fig. 1C, D). B-cell proportions remained stable in the response group, whereas they significantly changed in the non-response group (P = 0.0599) (Fig. 1E). The proportions of NK cells and CD3+ T cells did not significantly differ between the groups (Fig. 1F, G). Although some immune cell subsets showed significant differences in proportion, these changes reflect cell quantity rather than functional state. In contrast, our focus is on the functional activity of Tregs, as revealed by gene expression profiles, underscoring the need for more precise methods to guide treatment decisions.

A total of 60 patients with locally advanced or metastatic NSCLC receiving PD-1/PD-L1-based immunotherapy were retrospectively enrolled based on predefined exclusion criteria. Patients were classified into response (n = 31) and non-response (n = 29) groups according to clinical treatment outcomes (A). Peripheral blood samples were collected before and after treatment to assess changes in lymphocyte subsets. No significant differences in Treg cell proportions were observed in either group (B). CD4+ and CD8+T cell counts significantly increased after treatment in the response group, but not in the non-response group (C, D). B cell counts remained unchanged in the response group but significantly decreased in the non-response group (E). NK cells and CD3+ T cells showed no significant changes in either group (F, G). *P < 0.05, **P < 0.01, ns = not significant.
Single-cell analysis of T cell subsets
To further investigate the functional heterogeneity of Tregs at the single-cell level, we performed comprehensive transcriptomic profiling following standardized quality control. After quality control based on cell features, mitochondrial gene expression, and cell proportions, 41,202 high-quality cells were retained for analysis. Gene expression across samples is summarized in Supplementary Fig. 1A, B. A strong correlation was observed between sequencing depth and total cellular transcripts (R = 0.96; Supplementary Fig. 1C). Variance analysis identified 2000 highly variable genes (Supplementary Fig. 1D), which were normalized and subjected to principal component analysis (PCA). Twenty principal components were selected based on statistical significance (p < 0.05) for downstream analysis (Supplementary Fig. 1E). This study included seven samples with stable cell distributions and no significant batch effects; these samples were considered suitable for further analysis (Supplementary Fig. 1F). The core cells were classified into 16 independent clusters by the t-SNE algorithm, and a literature review confirmed the marker genes for each cluster, which ultimately categorized these clusters into nine groups: B cells, adipocytes, myeloid cells, neutrophils, plasma cells, T cells, endothelial cells, epithelial cells, and fibroblasts (Supplementary Fig. 1G, Fig. 2A, B). A total of 4,620 differentially expressed genes were identified between these cell clusters. The bar graphs show the numbers and proportions of different cell subtypes in each patient and the expression of marker genes in different cell types (Supplementary Fig. 1H).

Unsupervised clustering identified 16 cell clusters, grouped into nine major cell populations(A). Marker gene expression was used to annotate cell types(B). T cells were classified into Tfh, cytotoxic T cells, double-negative T cells (DNT), naïve T cells, natural killer (NK) cells and Tregs(C). Representative marker genes for each T cell subtype are shown(D). Tregs were subdivided into two transcriptionally distinct subclusters(E). Pseudotime trajectory analysis revealed a dendritic differentiation structure and identified the starting point(F). Three states were observed along the trajectory: pre-branch and two terminal branches(G). Treg cluster C1 was mainly located at the end of branch 2; C2 was enriched at the end of branch 1(H). Dynamic expression of differentially expressed genes along pseudotime was visualized as heatmaps(I).
Recent studies have emphasized the crucial role of intercellular communication in the progression of various cancers. For this reason, we analyzed the intercellular communication among cell subpopulations within the TME. In our study, strong communication was observed between immune and nonimmune cells (Supplementary Fig. 1L). Supplementary Fig. 1M shows the number of receptor pairs between T cells and other cell types. Specifically, through the CD99–CD99 receptor pair, T cells communicate with endothelial cells, epithelial cells, fibroblasts, adipocytes, and myeloid cells (Supplementary Fig. 1N). These findings suggest that the unique TME of NSCLC may involve specific cell–cell interactions. Following the methods described, we re-clustered and grouped 14,006 T cells, annotating these clusters on the basis of the expression of marker genes. Ultimately, we subdivided the T cells into Tfh, cytotoxic, DNT, naïve, NK, and Treg cells (Fig. 2C, Supplementary Fig. 1I, J). Key marker genes for each cell type are clearly displayed in bubble charts (Fig. 2D). Additionally, bar graphs were generated to provide detailed information on the quantity and proportion of each subtype of cells in each patient (Supplementary Fig. 1K).
To better understand Treg heterogeneity, we performed pseudotime analysis and identified two distinct Treg subtypes (Supplementary Fig. 1O, Fig. 2E). Through pseudotime trajectory analysis of these two Treg cell clusters, we established a map with branching points for different differentiation trajectories and identified the starting point of cell differentiation (Fig. 2F). With the progression of pseudotime, the cells presented three states, exhibited by the starting point of branching (the pre-branch state) and the two other branches (Fate1 and Fate2; Fig. 2G). Cluster C1 Tregs predominantly appeared at the end of trajectory branch 2, whereas Cluster C2 cells mainly appeared at the end of trajectory branch 1 (Fig. 2H). For further analysis, we studied genes whose expression significantly changed between branches (q-value < 0.01) and displayed the dynamics of these genes in Fig. 2I. A scatter plot was generated to highlight the six genes with the most significant changes (Supplementary Fig. 1P).
Identification of prognostic treg metabolism-associated genes via WGCNA
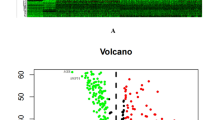
Given the critical role of Tregs in tumour progression, understanding the relationship between the infiltration of Tregs in NSCLC patients and their clinical relevance is particularly important. We used the Quantiseq algorithm to calculate Treg infiltration scores in cancer tissues and adjacent tissues of patients with NSCLC in the TCGA database. The analysis revealed that the Treg infiltration scores in the tumour tissue samples were significantly lower than those in the adjacent normal tissue samples (Fig. 3A). Patients were divided into high-score and low-score groups on the basis of the optimal cut-off value. K-M analysis revealed that the outcomes of patients in the high-score group were significantly better than those of patients in the low-score group (Fig. 3B). Although our results revealed a lower Treg infiltration score in tumour tissues, this may reflect a suppressive role of Tregs within the tumour microenvironment, suggesting that these cells may not perform their typical immunoregulatory function, which could have a complex impact on tumour prognosis19. On the basis of these results, WGCNA was used to identify genes associated with Treg infiltration scores in NSCLC. During the construction of the coexpression network, the soft threshold power β was 5 when the fit index of the scale-free topology reached 0.85 (Supplementary Fig. 2A). We statistically analysed the genes in the network and divided them into 22 modules, represented by different colours (Supplementary Fig. 2B). We found that the brown (cor = 0.81, p < 0.0001), magenta (cor = 0.60, p < 0.0000), and gray60 (cor = 0.50, p < 0.0001) modules had the closest relationships with Treg infiltration scores (Fig. 3C). The gene importance of all three modules was significantly positively correlated with that of their module members (Supplementary Fig. 2C). To further explore the relationship between Treg cell metabolism-related genes and the prognosis of NSCLC patients, differential expression analysis was performed on NSCLC and adjacent tissues. Under the screening criteria of FDR < 0.05 and |log2FC | > 0.585, 5000 genes were considered differentially expressed (Fig. 3D). Differential expression analysis was subsequently performed on tumour samples from patients with NSCLC with high and low Treg infiltration scores. Under the selection criteria of FDR < 0.05 and |log2FC | > 0.585, 1339 genes were considered differentially expressed (Fig. 3E). We intersected metabolism-related genes, genes differentially expressed in Tregs, genes related to the WGCNA modules, genes differentially expressed between tumour and adjacent tissues, and genes differentially expressed between the high- and low-Treg infiltration score groups to reveal 26 key Treg cell metabolism-related genes (Fig. 3F). These genes were selected for further investigations.

The significant difference in Treg infiltration scores between the paracancer samples and NSCLC samples are shown by the box - plot (A), and based on this, Kaplan -Meier survival curve demonstrated a difference in prognosis between groups with high and low Treg infiltration scores (B). The module genes were statistically analysed and divided into 22 modules, and it was found that the modules brown, magenta and grey60 had the closest relationships with Treg infiltration scores (C). Volcano map of differentially expressed genes in NSCLC and paracancer samples (D), and between groups with high and low Treg infiltration scores (E). Identifying 26 key Treg cell metabolic genes by intersecting different genes (F).
Construction and Validation of a Treg-Associated Prognostic Model in NSCLC
We conducted a univariate Cox regression analysis of the expression profiles of 26 genes to assess their potential impact on overall survival (OS) in patients. To minimize the possibility of omitting significant genes, we set the threshold for the p value as 0.120. The analysis revealed seven genes significantly associated with OS (Fig. 4A). Using the LASSO Cox regression method, we subsequently developed a prognostic feature model based on seven genes (BIRC3, TIMP1, G0S2, ACP5, PRKCB, PDE4B and CD52) (Fig. 4B–D). To evaluate the prognostic value of this model, we first stratified patients into high-risk and low-risk groups on the basis of median risk scores calculated by the model. Survival analysis revealed that the OS of patients in the high-risk group was significantly lower than that of patients in the low-risk group (Fig. 4E). Time-dependent ROC curve analysis also confirmed the model’s moderate accuracy in predicting 1-year, 3-year, and 5-year survival rates, with areas under the curve (AUCs) of 0.608, 0.640, and 0.622, respectively (Fig. 4F). Univariate and multivariate Cox regression analyses additionally revealed that, compared with other common clinical features, the risk score could independently predict the survival outcomes of NSCLC patients (Fig. 4G). Additional validation analysis demonstrated that the risk score model trained on the TCGA dataset exhibited consistent prognostic efficacy across multiple validation cohorts, with low-risk group patients showing a clear survival advantage (Supplementary Fig. 3A–E). To explore the potential application of this risk model in clinical practice, we integrated age, disease stage and the risk score into a nomogram to predict the overall survival of NSCLC patients. In this model, the higher the total score a patient receives on the basis of their clinical parameters, the poorer their survival prognosis (Fig. 4H). The calibration plots for the 1-year, 3-year, and 5-year survival revealed a high degree of conformity between the model predictions and actual observations (Fig. 4I). ROC curves further validated the accuracy of the nomogram in predicting clinical outcomes (Supplementary Fig. 4A), and decision curve analysis demonstrated its high clinical utility (Supplementary Fig. 4B). Finally, we analysed the relationships between risk scores and clinical features and found that the high-risk score group had a greater proportion of patients with advanced-stage disease and more instances of EGFR mutations and EML4-ALK fusion genes (Supplementary Fig. 4C).

A univariate Cox regression analysis was performed on the expression profiles of 26 genes to evaluate their potential impact on the overall survival (OS) of patients, and seven genes were significantly associated with OS (A). Subsequently, the LASSO Cox regression method was used to develop a prognostic feature model based on these seven genes (BIRC3, TIMP1, G0S2, ACP5, PRKCB, PDE4B, CD52) (B–D). Patients were stratified into high-risk and low-risk groups based on median risk scores calculated by the model, and survival analysis revealed that the OS of patients in the high-risk group was significantly lower than that in the low-risk group (E). Time-dependent ROC curve analysis confirmed the model’s moderate accuracy in predicting 1-year, 3-year, and 5-year survival rates (F). Univariate and multivariate Cox regression analyses identified independent prognostic factors, presented in a forest plot where the final column displays the hazard ratio (HR), 95% confidence interval (CI), and p-value in a compact format (G). A nomogram was established based on independent prognostic factors to predict overall survival (H), and the calibration curves for the nomogram are presented (I).
Immune microenvironment profiling and immunotherapy response prediction based on the prognostic model
To explore the relationship between the prognostic model risk score and immune status, we analyzed the differences in the expression of various immune modulators and types of immune cells in the high- and low-risk score groups. The results revealed that the expression levels of most immune modulators and immune cells were increased in the low-risk score group (Supplementary Fig. 5A, Fig. 5A). Considering the significant impact of the abnormal expression and function of immune checkpoint molecules in cancer immunotherapy, we further evaluated the correlation between the prognostic model and the expression of immune checkpoint molecules. The analysis revealed a significant correlation between most immune checkpoint genes and genes related to the prognostic model (Fig. 5B). Additionally, to gain a deeper insight into the potential biological mechanisms of the prognostic model, we performed gene set enrichment analysis (GSEA). KEGG pathway analysis revealed significant activation of pathways such as extracellular matrix receptor interaction, focal adhesion, and signalling pathways (e.g., the IL-17, PI3K-AKT and TNF pathways) in the high-risk group (Fig. 5C). Moreover, GO analysis by GSEA indicated significant activation of biological processes such as extracellular matrix adhesion, the epidermal growth factor receptor signalling pathway, the ERBB signalling pathway, and the extrinsic apoptotic signalling pathway in the high-risk score group (Fig. 5D). These findings provide deeper insights into the link between risk scoring and the biological behaviour of tumours.

Expression differences of immune modulators between high and low risk score groups (A), and expression correlations between genes characterising risk scores and immune checkpoints (B). Results of GSEA KEGG analysis and GSEA GO analysis in the high-risk score group (C, D). Differences in predictors of immunotherapy response between high and low risk score groups (E-H). Relationship between risk scores and immunotherapy response in the GSE126044 dataset (I, J).
We next examined whether the model could predict immunotherapy response. Compared to the high-risk group, low-risk patients had lower TIDE scores and higher MSI, CYT and TLS scores—factors associated with better response (Fig. 5E–H). These findings preliminarily suggest that a low-risk score is associated with a favourable immunotherapy response. To validate these findings, we further analysed the relationship between the risk score and the immunotherapy response in the anti-PD-1/L1 immunotherapy cohort by analysing the GSE126044 dataset. The analysis indicated that in patients who responded well to immunotherapy, the risk scores were significantly lower than those in patients who did not respond (Fig. 5I). Additionally, a greater proportion of patients in the low-risk score group responded to immunotherapy (Fig. 5J). These results support the potential value of the prognostic model risk score in predicting the immunotherapy response and suggest that our model may help identify patients who are likely to benefit from immunotherapy in a clinical setting.
Genomic alterations and chemotherapy sensitivity associated with the prognostic risk model in NSCLC
To explore the correlation between somatic mutations and our prognostic model, we analyzed single nucleotide variations (SNVs) in patients with NSCLC. The analysis revealed that patients in the high-risk score group presented higher mutation frequencies in genes such as KRAS, RP1L1 and ASTN1 than did those in the low-risk group (Fig. 6A). Additionally, we analyzed copy number variations and detected greater increases in copy numbers at certain chromosomal locations, such as chromosomes 7 and 11, in the high-risk group than in the low-risk group (Fig. 6B, C). These findings provide new insights into the relationship between risk score and tumour genetic heterogeneity. Furthermore, we explored the link between the prognostic model and drug sensitivity. Using the “oncoPredict” R package and drug data from the Genomics of Drug Sensitivity in Cancer (GDSC) project, we predicted the half-maximal inhibitory concentrations (IC50s) of drugs for NSCLC samples in the TCGA database. Through Spearman correlation analysis, we observed varying trends in the associations between the IC50 values of different chemotherapeutic drugs and risk scores. For example, as the risk score increased, the IC50 values for drugs such as BMS_536924, TANK_1366, linsitinib, JNJ38877605, refametinib, and PARP_9495 decreased (Supplementary Fig. 5B, Fig. 6D). Conversely, the IC50 values for GSK690693, PHA_665752, CD532, amuvatinib, venotoclax, and Ara-G increased with increasing risk score (Supplementary Fig. 5C, Fig. 6E). These results reveal potential correlations between the risk score and drug responsiveness, providing important evidence to facilitate the development of personalized drug treatment strategies.

Single nucleotide variations (SNVs) were compared between high- and low-risk NSCLC patients, with higher mutation frequencies observed in genes such as KRAS, RP1L1 and ASTN1 in the high-risk group (A). Copy number variation (CNV) analysis revealed more extensive amplifications in the high-risk group, especially at chromosomes 3q, 7p and 11q (B, C). Spearman correlation analysis showed that the IC50 values of BMS_536924, TANK_1366, linsitinib, JNJ38877605, refametini, and PARP_9495 were negatively correlated with risk scores (D), while the IC50 values of GSK690693, PHA_665752, CD532, amuvatinib, venetoclax and Ara-G were positively correlated (E).
Expression and clinical relevance of prognostic risk model–associated key genes in NSCLC
To further validate our predictive model, we selected key genes (TIMP1, BIRC3 and G0S2) identified through previous bioinformatics analysis that may have oncogenic potential in NSCLC. First, we validated the mRNA expression levels of these genes in cancerous and adjacent normal tissues via qRT‒PCR. The results revealed that TIMP1 and BIRC3 were significantly upregulated in tumour tissues (Fig. 7A, B), whereas G0S2 was more highly expressed in adjacent noncancerous tissues (Fig. 7C). Additionally, we correlated the mRNA expression levels of these genes with clinical and pathological data, as detailed in Supplementary Tables 2–4. To confirm these findings, we conducted immunohistochemistry (IHC) analysis on NSCLC tissue microarrays to assess the protein expression levels of TIMP1, BIRC3 and G0S2. The tissue microarrays included samples from 90 patients, but owing to some tissue sections detaching or showing high nonspecific staining, a portion of the patient data was excluded from the analysis (Supplementary Tables 5, 6). The IHC results indicated that TIMP1 and BIRC3 remained highly expressed in tumour tissues (Fig. 7D–G, Tables 1, 2). In contrast, G0S2 showed an opposite expression trend between RNA and protein levels: although its RNA expression was higher in adjacent non-tumourous tissues, its protein level was elevated in tumour tissues (Table 3, Fig. 7H, I). This discrepancy may be attributed to post-transcriptional regulation or protein stability, indicating that the complex regulation of G0S2 warrants further investigation21,22. On the basis of the IHC staining results, we classified patients into high and low expression groups for TIMP1 and G0S2, where “positive” and “strongly positive” staining were categorized as high expression, and “negative” or “weakly positive” staining were classified as low expression (Supplementary Fig. 6A, B). For BIRC3, since no negative staining was observed, patients were classified on the basis of staining intensity, with “weakly positive” indicating low expression and “positive” and “strongly positive” indicating high expression (Supplementary Fig. 6C). By correlating the expression levels of TIMP1, BIRC3 and G0S2 with clinical and pathological data, we found that all three genes were positively associated with various clinical factors. Specifically, high TIMP1 expression was positively correlated with clinical stage (Table 4, Supplementary Fig. 6D, E), high BIRC3 expression was strongly associated with lymph node metastasis and increased primary tumour size (Table 5, Supplementary Fig. 6F–I), and high G0S2 expression is correlated with increased lymph node metastasis. (Table 6, Supplementary Fig. 6J, K). Overall, these changes are closely related to cancer progression. Survival analysis revealed that TIMP1 expression was not significantly associated with overall survival (p > 0.05) (Fig. 7J), possibly because of the small number of patients in the low-expression group, making it difficult to reach a significant conclusion. The median survival time required for statistical analysis was not reached for the low BIRC3 expression group, indicating that the follow-up period was insufficient (Fig. 7K). The high G0S2 expression group had a significantly shorter median overall survival than the low G0S2 expression group did (Fig. 7L). Lung cancer tissue microarrays were stained using multiplex immunofluorescence (mIF) to investigate the relationship between BIRC3 and G0S2 and the development of NSCLC (Supplementary Fig. 7). Tregs were identified via colocalization analysis of CD4 and Foxp3, and the number of Tregs expressing the target molecules was statistically analysed via the Wilcoxon signed rank test. The results revealed that the number of Tregs expressing G0S2 and BIRC3 was significantly greater in cancer tissues than in paracancerous tissues (Fig. 7M–R). In conclusion, high expression of BIRC3 and G0S2 may serve as potential markers of a poor prognosis in NSCLC patients.

Using qRT-PCR to validate mRNA expression levels of TIMP1, BIRC3, and G0S2 in NSCLC tissues compared to adjacent non-cancerous tissues, with **P < 0.01, ***P < 0.001 (A-C). Quantitative analysis of TIMP1, BIRC3 and G0S2 expression in adjacent non-cancerous tissues and NSCLC tumour tissues. Statistical analysis was performed using the Student’s t-test, with ****P < 0.0001 (D–I). Kaplan-Meier survival curves for TIMP1, BIRC3 and G0S2 in NSCLC patients. Note: NA indicates not achieved (J–L). Multiplex immunofluorescence of tumour tissue (M, P) and adjacent tissue (N, Q), and CD4+, FOXP3+ and BIRC3+ ratio of tumour tissue and adjacent tissue (O, R).
G0S2 Overexpression Is Associated with NSCLC Progression and Increased Treg Infiltration
To investigate the association between G0S2 and a poor NSCLC prognosis, we opted for the murine NSCLC cell line LLC to establish a fully-immunocompetent mouse NSCLC model. We established an LLC cell line stably overexpressing G0S2, LLC-G0S2OE, along with an empty vector control cell line, LLC-EV-Control. Both cell lines were engineered to express luciferase for bioluminescence imaging. Western blotting revealed that endogenous G0S2 expression in parental LLC cells was below detectable limits, making this system ideal for gain-of-function studies (Fig. 8A, B). The uncropped and unprocessed scans of the blots are presented in Supplementary Fig. 8. Using these cell lines, we generated an orthotopic mouse lung cancer model. Survival analysis revealed that the survival of the mice in the G0S2OE group was significantly shorter than that of the control group, despite the lack of notable differences in body weight (Fig. 8C, D). To further confirm the role of G0S2 in lung cancer progression, we monitored tumour growth in the lungs of mice using small animal imaging to assess luciferase fluorescence signal. The results demonstrated that lung cancer progression was markedly accelerated in the G0S2OE group compared with the control group (Fig. 8E, F), which was corroborated by measurements of the tumour mass in the lungs collected post euthanasia (Fig. 8G). To evaluate whether G0S2 cells are associated with Treg infiltration in lung cancer, we performed flow cytometry on tumour tissue from the mice. While no significant differences in the infiltration of CD4+ T cells or CD8+ T cells (percentage of CD3+ T cells) were detected between the G0S2OE and control groups, Treg infiltration (percentage of CD4+ T cells) was significantly increased in the tumours of G0S2OE mice (P = 0.04) (Fig. 8H, I), which was consistent with the patient data. Additionally, no significant changes in Treg cell levels were detected in the spleen or peripheral blood of G0S2OE mice, nor were there differences in CD4+ or CD8+ T cell populations in these tissues (Supplementary Fig. 9). In summary, our findings in an orthotopic lung cancer mouse model demonstrate that G0S2 is associated with NSCLC progression and increased Treg infiltration.

Representative Western blot images showing G0S2 protein expression in LLC-G0S2OE (G0S2 overexpression) and LLC-EV-Control (empty vector control) cells. β-actin was used as a loading control (A, B). The models were established using the mouse NSCLC cell lines LLC-G0S2OE and LLC-EV-Control. Survival and body weight of mice bearing orthotopic lung cancer models were recorded. N = 7 (MST: median survival time) (C, D). Bioluminescence signals from orthotopic lung cancer models were monitored weekly using an in vivo imaging system. N = 4 (E, F). On day 28 post-tumour implantation, mice were euthanized, and lung tumours were excised and weighed. N = 5 (G). On day 28 post-tumour implantation, flow cytometry was performed to analyse CD4+ T cells, CD8+ T cells and Tregs in the lung tumour tissue. N = 3. All data are expressed as mean ± standard deviation. Statistical significance: ns, nonsignificant (P > 0.05); *, P < 0.05 (H, I).
Discussion
Recent advancements in understanding NSCLC heterogeneity and the TME have driven breakthroughs in improving long-term survival for patients with advanced disease. The shift from conventional chemotherapy to precision targeted therapy and immunotherapy has significantly improved patient outcomes. However, treatment efficacy remains limited due to tumour heterogeneity and immune escape mechanisms, emphasizing the need for effective biomarkers and therapeutic strategies.
Tregs play important roles in the progression and development of NSCLC by suppressing antitumour immunity and facilitating immune evasion. However, enhancing the efficacy of tumour immunotherapy and broadening its predictive value for immunotherapy remain key challenges. Metabolic molecules significantly influence the immune function of Tregs, which in turn impacts disease progression23. Thus, combining metabolic response indicators with the Treg cell immune status could offer a more effective approach to improve prognostic accuracy. However, it is important to note that only a subset of patients currently derive substantial benefits from tumour immunotherapy24. Increasing evidence suggests that the heterogeneity of the TME contributes to the variability in treatment outcomes25. The advent of scRNA-seq technology has revolutionized our understanding of the TME, enabling high-resolution analysis of intratumoural heterogeneity, cell differentiation trajectories, and intercellular communication26,27. This approach allows for a shift in focus from merely comparing tumour and normal tissues to examining the functional differences within tumour cells themselves28. In this study, we utilized scRNA-seq data to identify differentially expressed genes in Tregs. By integrating RNA-seq datasets with a metabolism-related gene set, we developed a seven-gene prognostic signature. This model was used to calculate risk scores and classify NSCLC patients into high-risk and low-risk groups. Survival analysis of the TCGA cohort revealed a significantly better prognosis in the low-risk group than in the high-risk group, and these findings were validated in the GEO cohort. ROC curve analysis further confirmed the model’s accuracy in predicting 1-, 3-, and 5-year survival outcomes. To our knowledge, this is the first study to explore the integration of tumour metabolism and immune-related prognostic features of Tregs in NSCLC. This approach not only enhances our understanding of the role of Tregs in the TME but also provides a valuable tool for stratifying patients and guiding personalized immunotherapy.
Notably, our analysis revealed that the metabolic prognostic feature consists of seven marker genes, including TIMP1, BIRC3, PRKCB, PDE4B, G0S2, CD52, and ACP5, and most of them were significantly overexpressed in high-risk groups, contributing to tumour progression and immunosuppression. Among these genes, we focused on G0S2 (G0/G1 switch gene 2) because its high expression in tumour tissues was associated with poor prognosis in our clinical cohort. G0S2 is emerging as a key regulator of cancer development and plays critical roles in lipid metabolism, apoptosis, and cell cycle regulation29,30. By inhibiting adipose triglyceride lipase (ATGL), G0S2 influences tumour metabolism, a hallmark of cancer, and promotes apoptosis through interaction with the antiapoptotic protein BCL2, suggesting its tumour-suppressive potential29,31,32. However, G0S2 plays dual roles, acting as a tumour suppressor in cancers such as breast and lung cancer, while contributing to tumour aggressiveness in other cancers, possibly due to its impact on metabolic regulation29. Its dysregulation is linked to tumour progression, therapy resistance, and adaptation to the hypoxic tumour microenvironment, making it a promising biomarker and therapeutic target. In our study, the identification of G0S2 as a biomarker underscores its dual role in regulating metabolism and Treg-mediated immune evasion, highlighting its potential as a therapeutic target. Notably, this was strongly and uniquely demonstrated for the first time through in vivo experiments in this study. Despite these advancements, there are limitations such as reliance on retrospective data and partial validation of the identified genes. Future studies should prioritize large-scale, prospective analyses and functional experiments to further elucidate the biological mechanisms underlying these findings and validate the role of G0S2 in NSCLC.
In conclusion, the integration of Treg metabolism-related genes into a robust prognostic model can significantly improve the response to NSCLC treatment by improving patient stratification and guiding personalized treatment decision making. The identification of G0S2 as a predictive biomarker and effective therapeutic target offers new avenues for the design of more effective immunotherapies. Additionally, the user-friendly nomogram developed in this study has good clinical applicability and provides a practical tool for risk-based treatment decision making and improved NSCLC management.
Methods
Clinical data
This study retrospectively collected data from 60 patients with pathologically confirmed locally advanced or metastatic NSCLC (Fig. 1A). These patients were treated at the Department of Oncology, First Affiliated Hospital of China Medical University, receiving PD-1/PD-L1-based systemic therapy as first-line treatment between July 2021 and December 2023. The inclusion criteria were: (1) NSCLC diagnosis based on cytological or histological evidence, with no EGFR, ALK or ROS1 driver mutations detected in patients with lung adenocarcinoma; (2) locally advanced or metastatic NSCLC (stages IIIB, IIIC or IV) according to the 8th edition of the AJCC cancer staging system; (3) complete baseline and imaging evaluation data available; (4) first-line treatment with immune checkpoint inhibitors (ICIs) in combination with another agent; and (5) good physical condition and adequate organ function, with an ECOG performance status of 0–2. All patients underwent regular follow-up for at least 6 months. Patients with neuroendocrine carcinoma, sarcomatoid carcinoma or those unsuitable for immunotherapy were excluded. The baseline characteristics of NSCLC patients were shown in Supplementary Table 1. This study adhered to the Declaration of Helsinki and was approved by the Ethics Committee of the First Affiliated Hospital of China Medical University (IRB Protocol No. 2022-272-2). Informed consent was obtained from all participants.
Data collection and preprocessing
We used the R package TCGAbiolinks to download gene expression profiles for lung adenocarcinoma (TCGA-LUSC), and performed a log2(FPKM + 1) transformation on the data. Subsequently, total survival time (OS) corrected by TCGA officials and clinical information of patients, including age, stage, gender, and grade, were obtained from an article published in the journal Cell. To validate our model, we also downloaded datasets GSE37745, GSE41271, GSE68465, GSE72094, and GSE157009 from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). When processing these GEO datasets, we converted probes to gene symbols based on the platform’s probe-to-gene mapping. Probes corresponding to multiple genes were removed; for multiple probes corresponding to the same gene, their expression values were averaged. We retained samples with complete clinical information from both TCGA and GEO data, specifically those with complete survival time and status. Additionally, we downloaded the GSE127465 dataset from the GEO database, which contains single-cell transcriptomic data of 7 NSCLC patients. We selected data from solid tumour tissues for further analysis. For the selection of metabolic genes, we identified 4,839 metabolic genes based on HALLMARK, KEGG, and Reactome databases.
Single-cell data processing
The R package Seurat (version 4.0.0) was employed for preprocessing single-cell RNA sequencing (scRNA-seq) data. Initially, to ensure data quality, the following criteria were set to exclude certain cells: (1) cells expressing fewer than 200 genes were removed; (2) cells with mitochondrial gene percentage over 20% were eliminated. Subsequently, the dataset was normalized using the “Normalize-Data” function, and 2000 highly variable genes were identified using the “FindVariableFeatures” function based on the “vst” algorithm. The data were then scaled and dimensionality reduction was performed using Principal Component Analysis (PCA), selecting the top 20 principal components for further analysis. The TSNE algorithm was utilized for data visualization and exploration, and cell clusters were identified using the k-nearest neighbors (KNN) algorithm and the “FindClusters” function with a resolution of 0.5. Cell clusters identified were annotated based on the research by Fengying Wu, Nayoung Kim et al. Additionally, differential expression genes between different cell types were identified using the “FindAllMarkers” feature in Seurat, setting min.pct = 0.25, logfc.threshold = 0.25, and retaining only genes with p-value < 0.05.
Cell communication
To investigate cell communication within NSCLC, we utilized the CellChat software package for analysis. CellChat includes databases of ligand-receptor interactions for humans and mice, enabling systematic inference of communication networks between cells by integrating scRNA-seq data with ligand-receptor interaction databases. Initially, we evaluated the main signalling inputs and outputs among all cell types using the CellChatDB.human database. Subsequently, by employing the netVisual_circle function, we depicted the strengths of intercellular communication networks from target cell clusters to various other cell clusters across all cell types. Finally, using the netVisual_bubble function, we generated a bubble plot that highlights significant ligand-receptor interactions between target cell groups and other cell groups within the tumour microenvironment (TME).
T-cell clustering analysis
For the subtyping analysis of T cells, we adopted a similar analytical approach, including the identification of highly variable genes, dimensionality reduction analysis, cell data integration using Harmony, and the identification of distinct cell groups through clustering analysis. To differentiate among the cell groups, we utilized the FindAllMarkers function in the Seurat package (min.pct = 0.25, logfc.threshold = 0.25), employing the default non-parametric Wilcoxon rank-sum test and Bonferroni correction method to identify differentially expressed genes (DEGs) between cell groups. Furthermore, we conducted biological process (BP) enrichment analysis and KEGG enrichment analysis on the DEGs in different cell groups using the clusterProfiler package, to gain deeper insight into their functional characteristics and potential biological pathways.
Pseudotime analysis
Single-cell pseudotime trajectory analysis was conducted using the Monocle2 package in R. The analysis utilized the raw UMI counts gene-cell matrix of Treg cells processed by Seurat as input data. Initially, a single-cell dataset object was created using the newCellDataSet function with the expressionFamily parameter set to negbinomial.size. Genes considered for trajectory analysis were those exhibiting differential expression across subtypes. Dimensionality reduction was then performed using the reduce Dimension function, with the method set to “DDRTree” and max_components set to 2. Cells were ordered and visualized using the plot_cell_trajectory function. To identify genes varying over pseudotime (q-Val < 0.01), these genes were sorted in descending order by their q-values. Their expression heatmaps were displayed using the plot_pseudotime_heatmap function, clustering genes into different subgroups based on their expression patterns.
Construction of Co-expression networks
Weighted Gene Co-expression Network Analysis (WGCNA) aims to identify modules of co-expressed genes, explore the association between these modules and specific phenotypes, and identify key genes within the network. Prior to conducting WGCNA, the infiltration level of Treg cells in NSCLC samples was assessed using the quantiseq algorithm within the IOBR package. Based on these scores, samples were divided into high and low infiltration groups using the optimal cut-off value. The survival time differences between these two groups of patients were compared using Kaplan-Meier (KM) survival curve analysis. The Treg infiltration scores for each sample were then used for WGCNA. In the WGCNA analysis, we focused on selecting genes with the top 50% coefficient of variation, ensuring that the genes analysed show significant expression differences in NSCLC. Initially, the correlation between any two genes was calculated, and these correlations were weighted to construct a gene network following a scale-free network distribution. Subsequently, a hierarchical clustering tree was constructed based on the correlations among genes. Different branches of the clustering tree represent different gene modules, each marked with a distinct colour. Afterwards, the significance of each module was assessed to determine the correlation between Treg cell infiltration scores and each module, and characteristic genes within each module were identified.
Construction of the prognostic model
Differential expression analysis between NSCLC and adjacent normal samples, as well as between high and low Treg cell infiltration score groups, was performed using limma. DEGs were selected by setting the threshold to an absolute log2 fold change ( | log2FC | ) > 0.585 and a false discovery rate (FDR) < 0.05. The intersection of metabolic-related genes, Treg cell-related differential expressed genes, WGCNA module characteristic genes, differential expressed genes between NSCLC and adjacent tissues, as well as differential expressed genes between high and low Treg cell infiltration score groups in NSCLC samples, were considered as key metabolic genes. Subsequently, key metabolic genes with prognostic significance were screened through univariate Cox regression analysis. To construct the prognostic model and reduce the risk of overfitting, LASSO penalized Cox regression analysis was applied using the “glmnet” R package for variable selection and shrinkage, thereby obtaining an interpretable model. The patient’s risk score was calculated based on the expression levels of each prognostic-related gene and their corresponding regression coefficients:
Note: n is the number of predictive genes, expo is the expression value of gene i, βi is the regression coefficient of gene i
Construction of nomograms
To integrate our risk score model results with clinical characteristics, we first constructed a nomogram in the TCGA NSCLC cohort using the “rms” R package, based on the independent prognostic factors identified through multivariate Cox regression analysis. This step aimed to create a comprehensive model that considers the interaction between the risk score and key clinical parameters. Subsequently, we plotted calibration curves to graphically demonstrate the consistency between actual survival rates and those predicted by the model, thereby assessing the model’s predictive accuracy. Furthermore, through decision curve analysis (DCA), we evaluated the net benefit of the model at different threshold probabilities to determine the practicality of the nomogram in clinical applications. This approach helped us quantify the predictive performance of the model, ensuring its value in real-world clinical decision-making.
Estimation of immune cell infiltration in the tumour microenvironment
We employed multiple algorithms integrated within the IOBR package, including cibersort, quantiseq, mcpcounter, estimate, and single-sample gene set enrichment analysis (ssGSEA), to quantitatively assess the relative abundance of various cell types within the tumour immune microenvironment (TIME) of patients with NSCLC. Based on the research by Charoentong et al., we used gene sets specifically marking different immune cell types in TIME, covering a variety of human immune cell subtypes including but not limited to activated CD8 T cells, activated dendritic cells, macrophages, natural killer T cells, and Tregs. Furthermore, by accessing the TISIDB database (http://cis.hku.hk/TISIDB/), we downloaded data on various immunoregulators (involving antigen presentation, cell adhesion, co-inhibitors, co-stimulators, ligands, and receptors). We compared the expression differences of different immunoregulators between high-risk and low-risk score groups and delved into their interactions and possible impacts on patient prognosis by calculating the Spearman correlation between genes in the risk model and immune checkpoints.
Potential Biological Mechanisms of the Prognostic Model
We focused on exploring the characteristics of the prognostic model for patients with NSCLC and its role in the response to immunotherapy, with a particular emphasis on the relationship with multiple immunotherapy predictive factors such as MSI, IFNG, TIDE, IPS, CYT, and TLS. MSI, IFNG, and TIDE for TCGA NSCLC were obtained through the TIDE database(http://tide.dfci.harvard.edu/). The IPS score was calculated using the IPS algorithm of the IOBR package, while the CYT score was defined by calculating the average expression of GZMA and PRF1. The TLS score was derived by applying the GSVA algorithm to a set of TLS characteristic genes. Additionally, to gain deeper insights into the impact of immunotherapy, we analysed NSCLC immunotherapy data from the GEO database, specifically patient progression-free survival (PFS) and treatment response in the GSE135222 and GSE126044 datasets. Risk scores were calculated for samples in these datasets, and survival differences between different risk score groups were analysed. Furthermore, through GSEA analysis, we compared pathway activity differences between different risk score groups. Analysis of hallmark pathway scores was conducted using GSVA and the limma package, selecting significantly different pathways with an FDR less than 0.05. Finally, somatic mutations (SNV) and copy number variations (CNV) data for NSCLC from the TCGA database were downloaded and analysed using GISTIC 2.0 and the TCGAbiolinks package to identify genomic variation characteristics of different risk score groups. Differences in SNV mutations between the two groups were compared using Fisher’s exact test.
Prediction of Chemotherapeutic Drug Sensitivity
To compare the differences in chemotherapy drug sensitivity between high-risk and low-risk scoring groups of NSCLC patients, we employed the oncoPredict algorithm. This algorithm, based on data from the Genomics of Drug Sensitivity in Cancer (GDSC) database, predicts the half-maximal inhibitory concentration (IC50) of drugs for TCGA NSCLC samples. We constructed a ridge regression model using the R package “oncoPredict” grounded on the drug data in the GDSC database, to predict drug sensitivity. Through Spearman correlation analysis, we explored the relationship between risk scores and drug IC50 values. To visually display the differences in drug sensitivity between different risk groups, we also plotted boxplots of the IC50 values for these drugs.
qRT-PCR
NSCLC and adjacent normal tissues were collected from the First Affiliated Hospital of China Medical University according to the following inclusion criteria: (1) Pathologically or cytologically confirmed resectable, treatment-naïve stage I-IIIA NSCLC (no restrictions on age or gender). (2) Eastern Cooperative Oncology Group (ECOG) performance status (PS) score of 0–1. (3) Signed informed consent form. (4) Complete baseline data, imaging assessments, and haematological specimens. Total RNA was extracted using the Trizol reagent kit (Shanghai Pufei, Cat. No. 3101-100) according to the manufacturer’s protocol. Reverse transcription was performed using the Promega M-MLV kit (Cat. No. M1705) following the manufacturer’s instructions. For qRT-PCR analysis, we employed the SYBR Master Mixture (TAKARA, Cat. No. DRR041B) to assess the expression levels of the target genes. The qRT-PCR thermocycling protocol was as follows: initial denaturation at 95 °C for 60 s, denaturation at 95 °C for 20 s, annealing at 55 °C for 20 s, extension 72 °C for 30 s, and amplification for 40 cycles. ACTB was used as the housekeeping gene. The primer sequences were shown in Supplementary Table 7. The 2-△△CT method was applied to calculate the expression level of genes and normalized to ACTB.
Immunohistochemistry
Fresh human tissue was fixed overnight in 10% formalin, dehydrated, paraffin-embedded, and sectioned. The sections were de-waxed, hydrated, and antigen-retrieved using citrate buffer. Peroxidase activity was blocked with 3% H2O2. Primary antibodies, including G0S2 (1:500, 12091-1-AP, Proteintech), BIRC3 (1:600, ab32059, Abcam), and TIMP1 (1:1000, ab211926, Abcam), were incubated overnight at 4 °C. Secondary antibody (from the Fuzhou Maixin UltraSensitive S-P Kit) was incubated at 37 °C for 1 h. DAB staining was then performed, followed by haematoxylin counterstaining. Sections were subsequently dehydrated, cleared, and mounted with neutral gum. Lung adenocarcinoma tissue microarrays were stained using IHC (Shanghai Xinchao Biotechnology Co., Ltd., No. HLugA180Su12). Staining was evaluated blindly without knowledge of the clinical information. Based on the IHC staining of tissue microarrays, patients with “positive” and “strong positive” staining were assigned to the high-expression group, while those with “negative” or “weak positive” staining were assigned to the low-expression group.
Multiplex staining and multispectral imaging
Four-color multiple fluorescence staining was performed using Opal series reagents according to the manufacturer’s instructions. Briefly, NSCLC tissue microarrays were deparaffinized with xylene and subsequently rehydrated through a series of different concentrations of ethanol (graded ethanol series). Antigen repair was then performed using a citric acid solution (pH 6.0) in a microwave oven. Endogenous peroxidase was blocked at room temperature and incubated for 10 min. This was followed by four consecutive staining procedures, which included incubation with primary and secondary antibodies and fluorescent labelling dyes, after again being blocked with goat serum for 30 min at room temperature. Finally, the nuclei were stained with DAPI and incubated for 10 min at room temperature. The samples were viewed and analysed using Slide Viewer software. Cell phenotypes were classified into the following subpopulations: G0S2+CD4+, G0S2+FOXP3+, BIRC3+CD4+, BIRC3+FOXP3+, CD4+FOXP3+G0S2+, CD4+FOXP3+BIRC3+. Tregs were identified by the co-localization of foxp3 and cd4. The target genes detected were G0S2 and BIRC3.
Cell lines and cell transfection
The mouse non-small cell lung cancer (NSCLC) LLC cell line was purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences. Cells were cultured in high-glucose DMEM complete medium containing 10% foetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C in a humidified incubator with 5% CO2. LLC cells in the logarithmic growth phase were seeded at a density of 3 × 104cells/well in 12-well plates and incubated for 24 h. Subsequently, lentiviral particles carrying the G0S2 overexpression plasmid were transfected into LLC cells using a transfection reagent. After 16 h, the medium was replaced with fresh complete medium. Transfection efficiency was evaluated 48 h post-transfection using fluorescence microscopy. Stable transfected clones (LLC-G0S2OE) were selected using puromycin treatment.
Experimental animals, grouping strategy, and lung cancer model establishment
Male C57BL/6 J mice (specific pathogen-free, 6–8 weeks old, 18–20 g) were purchased from Shanghai Slack Laboratory Animal Co., Ltd. and housed in specific pathogen-free (SPF) facilities at the Animal Centre of Shanghai University of Traditional Chinese Medicine, with environmental conditions maintained at 22 ± 2 °C, 55 ± 10% humidity, and a 12-hour light/dark cycle. All animal experiments were approved by the Institutional Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine (Approval No. PZSHUTCM220627040) and conducted in compliance with the ARRIVE guidelines.
Individual mice were randomly assigned to the LLC-G0S2OE group (n = 30) or LLC-EV control group (n = 30) using a computer-generated randomization sequence stratified by weight. Cage positions were rotated weekly to minimize environmental confounding variables. For tumour induction, LLC-G0S2OE and LLC-EV cells in logarithmic growth phase were resuspended in a 1:1 mixture of Matrigel and PBS at a final density of 1 × 105 cells/mL, with the suspension kept on ice until injection. Mice were anesthetized with 1.5% isoflurane (1 L/min oxygen flow) and maintained under continuous anaesthesia (1.0% isoflurane), monitored by pedal reflex and respiratory rate (40–60 breaths/min). A small incision was made along the left anterior axillary line to expose the thoracic cavity, and 100 μL of cell suspension was injected into the left lung via pleural puncture. Postoperative care included application of erythromycin antibiotic ointment and daily recovery monitoring.
Tumour progression was assessed weekly by intraperitoneal injection of D-luciferin (150 mg/kg), followed by bioluminescence imaging using an IVIS Spectrum system (PerkinElmer). Imaging began 15–30 min post-injection under 1.5% isoflurane anaesthesia, with signal intensity quantified as total flux (photons/sec/cm²/sr) using Living Image® 4.5.2 software. To ensure data validity, only animals with baseline flux ≥1 × 104 photons/sec/cm²/sr were included in longitudinal analyses, and no intraoperative mortality occurred (n = 0 excluded for surgical complications). Tumour cell inoculation (performed by researcher ML) and subsequent image acquisition/analysis (performed by researcher YB) were conducted under blinded conditions, with both investigators unaware of group assignments throughout the study. Humane endpoints were strictly implemented, including ≥15% weight loss, dyspnoea, persistent hypothermia ( ≤ 34 °C), or moribund state. Euthanasia was performed via incremental isoflurane exposure: 5% for 5 min to achieve surgical anaesthesia (loss of pedal and corneal reflexes), followed by 8% for 15 min until cardiorespiratory arrest. Death was confirmed by the absence of heartbeat and corneal reflexes for ≥5 min before necropsy. Sample size calculation based on tumour bioluminescence flux at day 28 (LLC-EV: 7.28 ± 0.31 vs. LLC-G0S2OE: 8.07 ± 0.40, Cohen’s d = 2.21) using G*Power 3.1 (α = 0.05, power=0.9) indicated that n = 3 per group was sufficient to detect this effect size. Survival analysis followed the intention-to-treat principle, including all randomized animals (n = 7/group), while bioluminescence dynamics and flow cytometry data were derived from mice surviving to the 28-day endpoint with complete measurements.
Flow cytometry
Lung tumours, spleens, and peripheral blood from mice were collected to prepare single-cell suspensions. Red blood cells were lysed in the dark for 5 min, followed by two washes with PBS. The cell concentration was adjusted to 1 × 107 cells/ml. Surface markers CD4, CD8, and CD25 were stained using FITC-conjugated anti-mouse CD4 antibody, APC-conjugated anti-mouse CD8a antibody, and Brilliant Violet 605™ anti-mouse CD25 antibody (BioLegend, CAS: 100405, 100712, 102035), respectively, and incubated at room temperature in the dark for 30 min. Intracellular FOXP3 staining was performed after fixation and permeabilization using the True-Nuclear™ Transcription Factor Buffer Set (BioLegend, CAS: 424401). Cells were then incubated with Brilliant Violet 421™ anti-mouse FOXP3 antibody (BioLegend, CAS: 126419) at room temperature in the dark for 30 min. The samples were analysed for marker expression using the CytExpert software on the CytoFlex LX flow cytometer (Beckman Coulter).
Western blotting analysis
Total proteins were extracted from LLC-G0S2OE and LLC-EV-Control cells using RIPA lysis buffer, quantified by the BCA method, and denatured at 99 °C for 10 min. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and blocked with 5% skim milk. The membranes were incubated with anti-G0S2 primary antibody (1:1,000, 12091-1-AP, proteintech) at 4 °C overnight, followed by incubation with HRP-labelled secondary antibody (1:20,000, 111-005-003, Jackson ImmunoResearch) at room temperature for 1 h. After G0S2 was detected using ECL substrate, the antibodies were stripped for 30 min, washed with TBST, and then blocked again. The membranes were incubated again with anti-β-actin primary antibody (1:20,000, 66009-1-Ig, proteintech) at 4 °C overnight, followed by incubation with secondary antibody (1:20,000, 115-035-003, Jackson ImmunoResearch) and ECL detection. Signal intensity was quantified by ImageJ and normalized with β-actin for data analysis.
Statistical analysis
In our study, all data analyses were performed using the R language version 4.1.2. To evaluate differences in expression levels, cell infiltration percentages, and various biomarkers, we utilized two statistical testing methods. For comparisons between two groups of samples, the Wilcoxon rank-sum test was employed to assess their significant differences. For comparisons among multiple groups of samples, the Kruskal-Wallis test was used. In illustrating these analytical results through various charts, we denoted significance levels with specific markers: not significant (ns) for p-values > 0.05, * for p-values < 0.05, ** for p-values < 0.01, *** for p-values < 0.001, and **** for p-values < 0.0001. Additionally, survival curves for prognostic analyses were generated using the Kaplan-Meier method, and the log-rank test was utilized to determine the significance of differences between groups.
Data availability
The data used and/or analyzed in the current study are available from the corresponding authors on request.
Code availability
Analytic code supporting the findings of this study is available from the corresponding authors
Abbreviations
- ACP5:
-
Acid Phosphatase 5
- AJCC:
-
American Joint Committee on Cancer
- ALK:
-
Anaplastic Lymphoma Kinase
- BIRC3:
-
Baculoviral IAP Repeat-Containing 3
- CD52:
-
CAMPATH-1 antigen
- CNV:
-
Copy number variation
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CYT:
-
Cytolytic activity score
- DNT:
-
Double Negative T cells
- ECM:
-
Extracellular matrix
- ECOG:
-
Eastern Cooperative Oncology Group
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- FAK:
-
Focal adhesion kinase
- FDR:
-
False discovery rate
- G0S2:
-
G0/G1 Switch Gene 2
- IC50:
-
Half maximal inhibitory concentration
- ICIs:
-
Immune checkpoint inhibitors
- IHC:
-
Immunohistochemistry
- IPS:
-
Immunophenoscore
- KM:
-
Kaplan-Meier
- KNN:
-
k-Nearest Neighbors
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- mIF:
-
Multiplex Immunofluorescence
- MSI:
-
Microsatellite instability
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PCA:
-
Principal Component Analysis
- PD:
-
Progressive disease
- PDE4B:
-
Phosphodiesterase 4B
- PR:
-
Partial response
- PRKCB:
-
Protein Kinase C Beta
- qRT-PCR:
-
Quantitative polymerase chain reaction
- ROC:
-
Receiver Operating Characteristic
- ROS1:
-
Proto-oncogene c-ros Oncogene 1 Receptor Tyrosine Kinase
- RTKs:
-
Receptor tyrosine kinases
- scRNA-seq:
-
Single-cell RNA sequencing
- SD:
-
Stable disease
- SNV:
-
Single nucleotide variant
- Tfh:
-
Follicular Helper T cells
- TIDE:
-
Tumour Immune Dysfunction and Exclusion
- TIMP1:
-
Tissue Inhibitor of Metalloproteinase 1
- TKIs:
-
Tyrosine kinase inhibitors
- TLS:
-
Tertiary lymphoid structures
- TME:
-
Tumour microenvironment
- TNF:
-
Tumour necrosis factor
- Tregs:
-
Regulatory T cells
- TSNE:
-
t-distributed Stochastic Neighbor Embedding
- UMI:
-
Unique molecular identifier
- WGCNA:
-
Weighted Gene Co-expression Network Analysis
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Howlader, N. et al. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 383, 640–649 (2020).
Duma, N., Santana-Davila, R. & Molina, J. R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 94, 1623–1640 (2019).
Boumahdi, S. & de Sauvage, F. J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 19, 39–56 (2020).
Sawant, D. V. et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 20, 724–735 (2019).
Setoguchi, R., Hori, S., Takahashi, T. & Sakaguchi, S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 201, 723–735 (2005).
Allard, B., Longhi, M. S., Robson, S. C. & Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 276, 121–144 (2017).
Cheng, W., Kang, K., Zhao, A. & Wu, Y. Dual blockade immunotherapy targeting PD-1/PD-L1 and CTLA-4 in lung cancer. J. Hematol. Oncol. 17, 54 (2024).
Gambardella, V. et al. Safety and Antitumor Activity of a Novel aCD25 Treg Depleter RG6292 as a Single Agent and in Combination with Atezolizumab in Patients with Solid Tumors. Cancer Res Commun. 5, 422–432 (2025).
Horton, B. L. et al. Overcoming lung cancer immunotherapy resistance by combining nontoxic variants of IL-12 and IL-2. JCI Insight. 8. https://doi.org/10.1172/jci.insight.172728 (2023).
Bendell, J. et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunol. Immunother. 72, 2443–2458 (2023).
Schoenhals, J. E. et al. Anti-glucocorticoid-induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front Immunol. 9, 2170 (2018).
Zemmour, D. et al. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 19, 291–301 (2018).
Jia, S., Bode, A. M., Chen, X. & Luo, X. Unlocking the potential: Targeting metabolic pathways in the tumor microenvironment for Cancer therapy. Biochim. Biophys. Acta, Rev. Cancer 1879, 189166 (2024).
Watson, M. J. et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651 (2021).
Ding, R. et al. Lactate modulates RNA splicing to promote CTLA-4 expression in tumor-infiltrating regulatory T cells. Immunity 57, 528–540.e526 (2024).
Field, C. S. et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 31, 422–437.e425 (2020).
Yang, D., Ma, X. & Song, P. A prognostic model of non small cell lung cancer based on TCGA and ImmPort databases. Sci. Rep. 12, 437 (2022).
Wolf, D., Sopper, S., Pircher, A., Gastl, G. & Wolf, A. M. Treg(s) in Cancer: Friends or Foe?. J. Cell. Physiol. 230, 2598–2605 (2015).
Zou, Y. et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int J. Surg. 107, 106936 (2022).
Jiang, D., Cope, A. L., Zhang, J. & Pennell, M. On the Decoupling of Evolutionary Changes in mRNA and Protein Levels. Mol. Biol. Evol. 40. https://doi.org/10.1093/molbev/msad169 (2023).
Dykema, A. G. et al. Lung tumor-infiltrating T(reg) have divergent transcriptional profiles and function linked to checkpoint blockade response. Sci. Immunol. 8, eadg1487 (2023).
Soltani, M., Zhao, Y., Xia, Z., Ganjalikhani Hakemi, M. & Bazhin, A. V. The Importance of Cellular Metabolic Pathways in Pathogenesis and Selective Treatments of Hematological Malignancies. Front. Oncol. 11, 767026 (2021).
Whiteside, T. L., Demaria, S., Rodriguez-Ruiz, M. E., Zarour, H. M. & Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 22, 1845–1855 (2016).
Tang, H., Qiao, J. & Fu, Y. X. Immunotherapy and tumor microenvironment. Cancer Lett. 370, 85–90 (2016).
Lambrechts, D. et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018).
Lim, B., Lin, Y. & Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer cell. 37, 456–470 (2020).
Huang, B., Han, W., Sheng, Z. F. & Shen, G. L. Identification of immune-related biomarkers associated with tumorigenesis and prognosis in cutaneous melanoma patients. Cancer cell Int. 20, 195 (2020).
Cremer, J. et al. Acidosis-induced regulation of adipocyte G0S2 promotes crosstalk between adipocytes and breast cancer cells as well as tumor progression. Cancer Lett. 569, 216306 (2023).
Gonzalez, M. A. et al. Loss of G0/G1 switch gene 2 (G0S2) promotes disease progression and drug resistance in chronic myeloid leukaemia (CML) by disrupting glycerophospholipid metabolism. Clin. Transl. Med. 12, e1146 (2022).
Zagani, R., El-Assaad, W., Gamache, I. & Teodoro, J. G. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 6, 28282–28295 (2015).
Yim, C. Y. et al. G0S2 Suppresses Oncogenic Transformation by Repressing a MYC-Regulated Transcriptional Program. Cancer Res. 76, 1204–1213 (2016).
Acknowledgements
We would like to express our gratitude to all the patients who participated in this study. This work was supported by the Joint Science and Technology Plan of Liaoning Province (Project No. 2023JH2/101700110).
Author information
Authors and Affiliations
Contributions
The last author L.S. is responsible for project design and guidance. M.Z., M.L., Y.H., and Z.X. are responsible for manuscript writing, literature retrieval, data analysis, clinical sample collection and experimental manipulation. Co-correspondence authors Y.B. and L.S. are responsible for animal experimental design, guidance and data analysis, while L.C. is involved in manuscript revision and formatting.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors agreed to publish this manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, M., Liu, M., Hu, Y. et al. G0S2: a potential target for NSCLC identified through prognostic models from multi-Omic analysis of regulatory T cell metabolic genes. npj Precis. Onc. 9, 198 (2025). https://doi.org/10.1038/s41698-025-00995-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-00995-6
