
Abstract
The sophisticated envelope of Gram-negative bacteria modulates the uptake of small molecules in a side-chain-sensitive manner. Despite intensive theoretical and experimental investigations, a general set of pathways underpinning antibiotic uptake has not been identified. This manuscript discusses the passive influx versus active efflux of antibiotics, considering the responsible membrane proteins and the transported molecules. Recent methods have analyzed drug transport across the bacterial membrane in order to understand their activity. The combination of in vitro, in cellulo and in silico methods shed light on the key, mainly electrostatic, interactions between the molecule surface, porins and transporters during permeation. A key factor is the relationship between the dose of an active compound near its target and its antibacterial activity during the critical early window. Today, methodology breakthroughs provide fruitful tools to precisely dissect drug transport, identify key steps in drug resistance associated with membrane impermeability and efflux, and highlight key parameters to generate more effective drugs.
Similar content being viewed by others
Introduction
A worrying group of pathogens has emerged from the Enterobacteriacae family, as reported by WHO (World Health Organization) and ECDC (European Centre for Disease prevention and Control) (ESKAPEE group)1. These Gram-negative bacteria possess a sophisticated envelope with two membranes which enables them to resist antibiotics by regulating the internal concentration of antibiotics2,3. Two antagonistic transports, permeation and efflux, constitute the primary mechanisms involved in resistance, which are termed “the membrane-associated mechanism of resistance” (Fig. 1). This contributes to the susceptibility of bacteria towards antibiotics4,5. Several reports have described the involvement of membrane impermeability in clinical isolates, in terms of alterations in the expression of porins and efflux pump due to complex regulatory mechanisms that monitor their levels during drug treatments2,6. Concurrently, experimental and computational studies have investigated the role of antibiotic structure during its permeation across membrane channels such as porins and efflux transporters2,4,7,8,9,10,11,12.

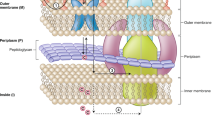
The blue and red arrows illustrate the penetration and expulsion fluxes, respectively. Steps 1, 2, 3 of influx (blue) and 1, 2, 3 of efflux (red) are detailed in the corresponding tables: During molecule travel across the bacterial membrane, within porins or efflux pumps, distinct key steps occur at distal, central and proximal locations/events. Distal step: This step involves the initial contact between the amino-acid side chains at the porin mouth and the charged groups of the molecule that pre-orientate the molecule or contribute to the molecule recognition. This is akin to the first contact between antibiotic chains and the PBP of the efflux pump. Second step: A strong electrostatic dialogue occurs between the molecule-specific site of the transporter, e.g. the eyelet region for porins and DBP for pumps. The residues lining the eyelet of the OmpF porin (Protein Data Bank ID 3K1B) that contribute to the electrostatic field are shown in a cartoon representation from an extracellular view. Proximal step: The final step involves the release of the molecule into the periplasmic space or through the TolC funnel, depending on the system. Each step involves energy thresholds defined by electrostatic interactions, electrostatic fields, conformational changes, antiport, membrane potential. OM and IM represent the outer membrane and the cytoplasmic (inner) membrane, respectively. “T” denotes the treatment administered to the patient. “?A-?E” denote gaps in antibiotic values at various positions: “?A” In vivo drug concentration in the bacterial environment; “?B”, Concentration of periplasmic-targeted drug in the periplasm; “?B*” Relation between periplasmic active drug concentration and target mutations or drug-modifying enzymes; “?C” Concentration of cytoplasmic-targeted drug in the cytoplasm; “?C*” Relation between cytoplasmic active drug concentration and target mutations or drug-modifying enzymes; “?D” Drug translocation through porin, environment dependence; “?D*” Relation between drug translocation through porin and porin type; “?E” Drug translocation through the efflux pump; “?E*” Relation between drug translocation through the efflux pump and pump type. Created in BioRender. Vergalli, J. (2024) BioRender.com/l86d043.
Despite intensive efforts, data regarding antibiotic activity are inconsistent with bacterial susceptibility as measured by inhibitory diameter, minimum inhibitory concentration (MIC), or extrapolation from culture growth2,3. Until recently, analyzes measured the relationship between the structure of molecules and their antibacterial activity based on MIC values. However, these measurements are taken after long incubation times, despite different studies reported a rapid bacterial response to external stresses13,14. Such information, depending on multiple factors ranging from the external concentration of the antibiotic to the observation/detection of inhibition, remains interpretive. This general protocol did not account for intermediate steps such as penetration, accumulation, susceptibility to efflux pumps, etc., relying instead on an overall view without defining the molecular and kinetic specificity of these key steps. This creates a scientific gap and a critical challenge for clinicians due to the uncertainty about the intra-bacterial concentration required for efficient antibacterial action, considering the possible role of target mutation and/or drug alteration by bacterial enzymes. It is therefore crucial to gain a deeper understanding of the dialogue between the molecule and the transporters. This will not only advance our knowledge of molecule diffusion across biological membranes, but also inform rational drug treatment relevant to bacterial phenotype and drive the urgent research of new series of pharmaceutical compounds required in infectious disease.
Over the last decade, new methods and concepts have emerged to identify and quantify small molecules in bacteria (live or lysed) as a function of incubation time, thanks to the development of fluorimetry and mass spectrometry technologies. Additionally, new tools are available to monitor bacterial physiology and detect events altering normal growth at an early stage. Finally, the increased computer power and efficiency of algorithms have made in silico methods more reliable than ever at dissecting processes at the atomic level.
The combination of these advances offers the possibility of addressing and filling the various gaps regarding the concentration, localization, and activity of antibiotics during initial contacts with bacterial cells. This manuscript presents the potentials offered by recent methods and concepts and discusses the future perspectives opened by these relevant applications.
In and out transport: key milestones, evidences and significant gaps
Bacteria have evolved various mechanisms of resistance to evade the inhibitory or killing effects of antibiotics by preventing the entry of antibiotics, actively extruding antibiotics outside the cell, degrading or modifying antibiotic molecules, and protecting or modifying cellular targets. The bacterial membrane is the first and most effective line of defense when faced with an antimicrobial drug. It can manage the rate of drug penetration while activating efflux pumps that push the drug out of the cell. This initial defense mechanism limits the internal drug concentration and allows bacteria to activate and/or acquire additional resistance mechanisms, thereby increasing its level of resistance. Both fluxes, influx through porins and efflux via efflux pumps, involve key steps where passive and active transporters and the drug subtly interact (Fig. 1).
During influx, the drug is pre-positioned by the charges of the porin channel and the ionized groups on its surface and the molecule will be oriented in the electrostatic field inside the pore3. The dipole alignment must be strong enough to pass the steric barrier of the eyelet, where the electric field involving residues of loop 3/anti-loop 3 is most intense. Diffusion rate then depends on a balance between spatial orientation and electrostatic interactions (Fig. 1) as recently proposed3,15,16,17.
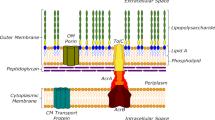
About the efflux system, the tripartite RND (Resistance-Nodulation-Division) pumps are widespread among Gram negative bacteria and often associated with multidrug resistance18,19. Upon efflux through the paradigmatic RND system of Escherichia coli, AcrAB-TolC, the substrate binds to the Proximal Binding Pocket (PBP) of AcrB, inducing conformational changes that shifts the protomer from the loose state (L) to the tight state (T) (Fig. 1). The substrate then moves and binds to the residues of the Deep Binding Pocket (DBP). Proton translocation, promoted by conformational changes in AcrB, ensures the transition of the protomer from the T state to the open state (O), allowing the released substrate to exit through the funnel formed by the AcrA-TolC complex.
It is evident that there are similarities between drug fluxes through porins and efflux pumps (Fig. 1). Both are governed by subtle interaction balance, in which electrostatic interactions between drug residues and the porin/efflux pump play a key role. Reorientation within the confinement area occurs in both the porin and the AcrB pump. Both transports also depend on membrane fluidity, interactions between the transporter’s monomers, the behavior of the solvent in porins and pumps, the charge of the transporter channel/pocket, and the charge and flexibility of the drug3,4,18.
This information clearly shows the similarities occurring during these transports, both of which are subject to go/no go steps (Fig. 1). These steps settle the drug concentration close to its target in real-time. This resident time concentration close to its target (RTC2T)2 determines the bacterial fate: cell death if the concentration is sufficient, or survival and potential acquisition of additional resistance mechanisms if the threshold is not reached4.
There are several key gaps in our understanding of antibiotic transport through porins and RND pumps (see “A-E” points described in Fig. 1). Investigating and dissecting these gaps will enable the determination of:
- the mechanics of transport and the kinetic parameters governing transport through porins and efflux pumps,
- the molecular interactions between the drug and the transporter during key stages of transport,
- the relationship between internal accumulation and antibacterial action during initial contact.
The final objective is to optimize the design of drugs whose structure will promote their rate of diffusion and accumulation near the target and to provide clinicians with a valuable tool for making informed choices about the best molecules to use.
Understanding the drug transport across membrane with different models
The study of drug transport through bacterial membranes is best served by combining parameters extracted from models at different scales: the whole bacterial cell, single transporter or channel, and the molecular interactions within the protein. Numerous in vitro, in cellulo, and in silico methods have been developed over the past few years, leading to significant advances in understanding and establishing mathematical models of the kinetics of drug flux across the membrane of Gram-negative bacteria20,21,22. From a non-exhaustive list of methods, we highlight and discuss their respective characteristics, advantages and disadvantages, taking into account their feasibility and their relevance for studying the transport and accumulation of antibiotics in bacterial cells (Fig. 2 and Table 1). To achieve the best results, “transportomic” methodologies must meet the following criteria: a short time scale, unmodified drug and transporter and, when possible, transporters in their natural environment (membrane integrity, affinity for compounds target). Achieving this latter point requires the development and use of in cellulo based methods, while kinetics parameters and molecular aspects can be respectively investigated by in vitro and molecular scale models. These findings should then be integrated into a global perspective approach, as discussed in the Conclusion (Fig. 3).

For in cellulo methods, the pros (green) and cons (red) of each method are illustrated in a circular heat map chart across the following categories: Transport: Relevant method for studying influx + efflux versus multi-factorial processes, Early time: Method applicability to early versus prolonged bacterial-drug contact, kinetics: Kinetic data versus discrete time points, True drug: Use of unmodified and active drug versus modified drug (labeled) and/or substrate without antibacterial activity (dye), quantitative: Provision of quantitative versus qualitative-relative data, All drugs: Applicability to all drugs versus specific drugs (fluorescent, same antibiotic family), single-cell: Data at the single-cell versus population level, sub-cellular: Ability to enable or not sub-cellular localization. The pros and cons of in vitro and in silico methods are also indicated in green and red, respectively. A.I. Artificial intelligence, DUV Deep UV, EOF Electroosmotic Flow, FARMA Fluorescent Artificial Receptor-based Membrane Assay, LUV Large Unilamellar Vesicles, MD Molecular Dynamics, MIC Minimal Inhibitory Concentration, Tof-SIMS Time-of-Flight Secondary Ion Mass Spectrometry. Created in BioRender. Vergalli, J. (2024) BioRender.com/v13n890.

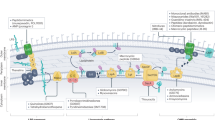
Red circles indicate sampling ___location (biopsy or collection) that are subjected to environmental conditions (green circle). Black text denotes the different proposed analyses to determine bacterial and molecules parameters. Different colors represent the respective methods and tools used during parameters analyses. Blue text indicates the recovery and analyses of parameters related to bacteria and molecules, with AI being used to highlight key points and specific insights. The analyses of the collected data pave the way for developing adapted animal models that consider the various identified parameters (e.g. local concentrations, combinations…). ATB: Antibiotic, MAMR: Membrane-associated mechanisms of resistance. “!A- !E” correspond to the response to the ”?A-?E” gaps mentioned in Fig. 1: - “!A”: In vivo drug concentration at infection site; - “!B”: Periplasmic active drug concentration determined in the absence or presence of an enzyme inhibitor, membrane permeabilizer, with/without target mutation, or using multiple combinations; - “!C”: Cytoplasmic active drug concentration determined in the absence or presence of an enzyme inhibitor or membrane permeabilizer, with/without target mutation, or using multiple combinations; -“!D”: Drug translocation through porin determined in the presence or absence of a membrane permeabilizer and relationship with the type of porin determined in the presence of identified porin; -“!E”: Drug translocation through efflux pump determined in the absence or presence of an efflux inhibitor/blocker and relationship with the type of efflux pump determined in the presence of identified pumps (e.g. AcrAB, AcrEF, ..etc). ‘T’ represents the formatting of information that can be used by clinicians during the selection of antibiotics or drug combination, or by pharmacochemists for the rational synthesis of new molecules. Finally, new strategies to counter MAMR are to be implemented to enhance clinical effectiveness. Created in BioRender. Vergalli, J. (2024) BioRender.com/h37t256.
In cellulo models
Most assays commonly employed in microbiology to assess bacterial antibiotic resistance are unsuitable for studying membrane transport (Fig. 2, Table 1). However, activity measurements on isogenic strains engineered to express varying levels of efflux pumps or porins can provide valuable insights into drug susceptibility to these membrane barriers23. Additionally, an initial assessment of the potential involvement of permeability defects or efflux susceptibility in the observed phenotype can be made by determining the MICs of drugs in combination with an efflux inhibitor (e.g., PAβN) or a permeabilizer (e.g., PMBN)24,25,26.Nevertheless, it is important to approach interpretations of influx and efflux based on activity measurements with caution, as they not fully capture the complexities of membrane transport. These measurements are multifactorial (involving enzymatic resistance, target affinities, cell physiology state), too long in duration to measure a relatively fast transport27, making analysis difficult and potentially biased. Additionally, the use of adjuvants can induce adverse effects and should be carried out with particular care, using largely sub-inhibitory concentrations. For example, PAβN, exhibits a permeabilizing effect on the bacterial membrane28,29. Similarly, the use of permeabilizers can lead to envelope destabilization, which may vary depending on the strains studied and affect membrane energy. Finally, it should be noted that the use of efflux pump substrates as “inhibitors” depends on their interactions with transporters and may not necessarily induce specific competition with the substrate at the same transporter site30,31.
Methods based on the use of fluorescent compounds or dyes (efflux pump substrates, markers of membrane destabilization, metabolic activity, cell viability or mortality, etc.) have boosted our understanding of the parameters involved in drug transport in bacteria32,33. The main advantage of these methods lies in their ability to measure early-time effects when bacteria are first exposed to the compounds. For instance, “Real Time Efflux” assays have the advantage of monitoring the initial stage of efflux using fluorescent compounds known to be substrates of efflux pumps32. The resazurin-reduction-based antibiotic uptake assay allows comparison of the influx capacity of various drugs in Enterobacteriaceae strains expressing different types of porins34. This assay indicates differences between strains expressing different porins that were not detected in MIC data. Conversely, this assay should be avoided when investigating the efflux capacity of non-isogenic strains, as resazurin is a substrate for efflux pumps35. These assays have the disadvantages of being solely qualitative, and in some cases they only consider one of the two transport, e.g. efflux is studied without examining differences in influx.
To date, accumulations assays, which determine the intracellular concentration of the drug resulting from influx and efflux, appear to be the most accurate approaches for studying both transport processes. Using microscopy with synthetic fluorescent antibiotics allows for real-time visualization of their accumulation within cells36,37. Dyes have also revealed different accumulation patterns depending on the bacterial growth phase: efflux impacts the accumulation in exponentially growing cells but not in stationary-phase cells, where accumulation seems dependent on membrane permeability38. Fluorescent probes and dyes can also be valuable tools in the development of antibacterial agents. Interestingly, they can be used - to evaluate membrane permeabilization in Gram-negative bacteria using vancomycin-fluorescent conjugates which would not enter cells with an intact membrane39, or - to assess the ability of efflux pump inhibitors to increase intracellular accumulation40. Although very useful and having provided extensive knowledge in the field of drug membrane transport, the use of dyes or labeled antibiotics does not accurately account for the possible molecular interactions between an unmodified antibiotic and its transporter.
Accumulation methods based on fluorimetry or mass spectrometry offer the significant advantage of being quantitative, thus allowing inter-laboratory, inter-strain and inter-molecule comparisons, and enabling the measurement of the accumulation of unmodified antibacterial compounds, whether naturally fluorescent (fluorimetry) or not (mass spectrometry) (Fig. 2, Table 1). About the latter, having a high sensitivity, it allows the study of low drug concentrations41,42 and can be applied to a wide range of molecules because it is not limited to those requiring labelling. Fluorimetry assays benefit from robust controls associated with internal standards and allow for precise quantification of the drug content in bacteria43. These methods (see reviews43,44 for methodological aspects) are widely used to determine the characteristics of compounds most likely to accumulate in Gram-negative bacteria45,46,47 (see paragraph “In cellulo contributions to drug design”), -to investigate differences in compound susceptibility to influx and efflux12,30, - to quantify efflux rates in clinical isolates27, - to determine species-specific dependence on porin-mediated drug transport48, - to check the selectivity of cephalosporin transport based on the type of porin expressed34, and finally to provide guideline for converting anti-Gram-positive drugs into anti Gram-negative ones49,50,51. Among fluorimetry assays, deep ultraviolet microscopy provides access to almost real-time follow-up of drug fluxes inside the bacteria52. Notably, insights were gained regarding dose and time parameters in transport, which are pivotal during antibiotic administration as they influence the efficacy of the drug as well as the acquisition of resistance53,54. Early times points play a crucial role in drug transport, with a steady state of accumulation in Gram-negative cells typically occurring within the first 5-10 minutes as demonstrated by microspectrofluorimetry55. Furthermore, the study demonstrated the impact of drug dosage on efflux activity, which exhibits high levels at low doses and reaches saturation at higher doses42.
In addition, deep UV microscopy allow us to consider cell phenotype heterogeneity within a population52 that is masked by population-level approaches. Single-cells methods, using deep UV on intact antibiotic or microscopy with labeled antibiotics have revealed different accumulation levels in individual cells within a population55. For example, lower accumulation has been observed in rapidly growing phenotypic variants compared to slow-growing cells56. Flow cytometry, another single-cell method, though not quantitative and often requiring a strong fluorescence signal—hence the use of probes or dyes—also offers the advantage of revealing population heterogeneity40. These differences at the cell level are important for the population, as a subpopulation of tolerant cells with low accumulation could lead to heteroresistance profile57,58.
In vitro models
In vitro models, using a simplified bacteria envelope, are the optimal method for structural and biophysical characterization of membranes and membrane transporters. They provide a powerful complementary approach to studying drug transport at a molecular level59,60 (Fig. 2, Table 1). To reduce the complexity of antibiotic uptake, selected porins are isolated and reconstituted into liposomes. The liposome swelling assay definitively probes the concentration-driven influx through porins by mixing functionalised vesicles under isosmotic conditions61. This assay is an important tool for understanding the uptake of nutrients or antibiotics through porins and require caution when working with charged compounds. These early studies definitively established the molecular sieving properties of porins and provided an explanation for the high diffusion rates of these compounds through the outer membrane.
A concentration gradient of charged compounds creates a so-called diffusion potential caused by a possible difference in electrophoretic mobility. This approach is well known as a selectivity measurement but it has only recently been applied for a semi-quantitative flux estimation62. A single channel conductance measurement combined with translocation number of between 1 and 100 antibiotic molecules extrapolated to a concentration gradient of 1 mM has been obtained60. It should be noted that the cation’s permeation depends strongly on the choice of anion and vice versa. Therefore, the permeability is not a simple constant but varies with salt concentration and composition. Furthermore, the potential binding of divalent cations like Mg2+ to carboxyl groups into the channel might even reverse the selectivity.
A different approach is the recently developed FARMA (fluorescent artificial receptor-based membrane assay), which uses fluorophore displacement to monitor the permeation of antibiotics across isolated porins34,63. A donor-acceptor pair forming a fluorescent complex is encapsulated into liposomes. The donor-acceptor pair must have the property to dissociate in the presence of the antibiotic to be tested. The antibiotic is added to the external solution, and, if able to use porin channel to pass the lipid membrane, the fluorescence will change due to competitive binding of the antibiotic to the complex. The kinetics of the resulting fluorescence decrease provide a real-time measurement of antibiotic translocation through the porins. The quencher can also be aptamers of antibodies.
Another promising way for understanding transport was the introduction of artificial amino acids into the porin. However, the fluorescence labelling was not sufficient to allow single molecule detection64.
In the case of a binding site inside the constriction zone, single channel recording is the ideal method for studying permeation. The entry of the molecule of interest will reduce the ion current, which will be visible as a short ion current block. However, blocking alone does not allow us to conclude on permeation as it is obvious that the molecules can simply bounce back. To draw conclusions on transport, one must take advantage of the electroosmotic flow. Most channels are ion selective. By applying a transmembrane voltage, the mobile ion will move inside the channel, creating a strong flow that can drag uncharged molecules through the channel. Charged molecules might even flow against the electric field. Increasing the external voltage increases or decreases the residence time. The electroosmotic forces provide a strong flux, allowing you to distinguish permeation from simple blocking60,65,66.
By combining in cellulo and in vitro assays, the cephalosporins flux has been quantified in the two main classical E. coli porins OmpF and OmpC34. The results show a significant variation in the diffusion rate of ceftazidime vs cefepime/cefotaxime depending on the porin expressed. The variation in diffusion rates can be explained by a difference in the interaction between the antibiotic and the porin channel upstream the conserved restriction region termed “the eyelet”34. These results provide a possible explanation for the observations made on clinical strains showing a decrease in the expression of OmpF-like porins17. However, it is important to note that discrepancies are observed between the data collected by these different methods. For instance, a translocation of ~300 molecules per second of ceftazidime was measured though single OmpF channel in a planar lipid membrane, far above what is measured in cellulo34. This disagreement can be attributed to an extra barrier presented by the dense lipopolysaccharides layer, which reduces the overall permeability. Indeed, translocation measurements using porins inside outer membrane vesicles revealed lower permeabilities65. A second possibility is that some of the OmpF channels are in a closed state. However, no experimental evidence has been presented to support this hypothesis. The third most plausible explanation is that the periplasm is a crowded space in which a few molecules already have a high concentration. This example illustrates the importance of using different models to better understand the complexity of kinetics.
In silico models
Computational methods applied to bio-inspired questions and to medicinal chemistry have provided remarkable insights for the design of new therapeutic strategies67,68,69,70. These insights have been gained due to new techniques with different levels of accuracy and throughput, which are adaptable to the kind of questions to be tackled (Fig. 2, Table 1).
The advent of machine learning and deep learning methods have undoubtedly boosted the spectrum of applications of computational methods. The AI-driven combination of different in silico approaches and experimental techniques has enabled the re-direction of research and design of more efficient compounds45,71,72. Computational tools available to tackle diverse questions range from docking methods to more accurate quantum mechanical techniques. However, a trade-off between accuracy and outcomes is necessary. More accurate calculations can be restricted to smaller systems and/or a reduced number of cases, sampling shorter time scales with a lower throughput. More approximate techniques can handle large systems for long times or large sets of systems, creating large collections of data.
Homology modelling and beyond: structural insights
To exploit the structure-function paradigm, three-dimensional arrangements of biosystems, including receptors, ligands, lipids, etc., are essential. However, despite the impressive improvement of experimental techniques73,74, structural data are often missing, restricted to a specific configuration (apo or holo) or extracted in conditions different from the physiological ones and must be acquired computationally75. This is also necessary to provide structures for further investigations. AI has triggered an extraordinary breakthrough76,77, as evidenced by the appearance of AlphaFold78 and RoseTTaFold79. Even more impressive results based on AI have been achieved by the developments of AlphaFold, namely AlphaFold 278 and AlphaFold 380. By improving the structures of the computational protocols and the AI procedures, the two platforms have been able to predict the structures of protein complexes with DNA, RNA, and post-translational modifications. However, it is important to note that the predictions of these AI-derived techniques should be treated with caution. For example, they do not include the description of atomic positions for cofactors, metal ions, water molecules and any other ordered, bound ligands, although AlphaFold 3 is able to provide the structure of proteins in complex with some selected ligands and ions. Additionally, they are limited to the canonical isoform of a protein81,82, which can represent a limitation because different isoforms of the same proteins may have different structural arrangements and different functionality. This means that traditional homology modelling procedures can still be used to bridge some gaps left by the AI-based pipelines.
Specific to RND efflux systems, homology modelling83 was used to perform a thorough analysis of AcrD, a member of the RND family in E. coli84, of MexY, MexD, and MexF, RND transporters expressed in P. aeruginosa85,86, for which no structural data were available. The study of the obtained structures made accessible features, such as cavity volumes and channels, which cannot be extracted from the sequence analysis. Moreover, the 3D structure allows the evaluation of lipophilic index and electrostatic potential in different regions of the transporters, as resulting from the spatial arrangement of the residues. For instance, in the DBP of AcrD, the affinity site identified as the main hub of the RND transporters, there is a relatively dense positively charged environment compared to AcrB. This originates from the electrostatic contributions of amino acids like Arginine and Lysine, replacing their less polar counterparts in AcrB. The low lipophilicity of this pocket in AcrD, combined with the observed mosaic-like electrostatic patches provides a favourable binding site for anionic beta-lactams as well as for polycationic aminoglycosides. In contrast, the poor electrostatic and hydrophilic complementarity provided by the DBP in AcrB permits the binding of charged molecules like anionic beta-lactams but with far less affinity than that in AcrD.
Molecular docking
Docking is a precious tool in drug screening and the study of protein–protein interactions87,88,89. However, as with any approximated technique, problems can arise. These include inaccurate binding sites for target proteins, screening based on unsuitable small-molecule databases and inconsistency with bioassays90. Nevertheless, docking methods provide possible configurations of ligand-receptor and protein-protein complexes, which are essential for maximizing the starting positions of Molecular Dynamics (MD) simulations. This is demonstrated by the study of AcrB interacting with fluoroquinolones12,30, carboxylated drugs91 and cephalosporins92. In the case of the RND transporters, which are characterized by polyselective and large affinity sites (associated with the concept of the so-called diffusive binding), this way of exploiting the docking poses enables a sampling of different regions of the transporter and configurations of the ligands. Additionally, molecular docking-based virtual screening techniques are essential for screening out promising drug precursors from the vast amount of structural data available93.
Molecular dynamics simulations
MD simulations have demonstrated the ability to capture several microscopic details of biosystems94,95,96,97. Over the years, they have extended the meaning of the structure-function relationship by introducing the effects of dynamics at large, that is, the role of physiological-like conditions. This requires developing techniques that bridge the gap between biology and the in-silico realm while keeping an acceptable balance between accuracy and computational costs. Biased methods98,99,100,101 allow the sampling of events occurring on time scales not accessible to standard MD simulations. Two applications of these approaches to RND transporters have shown and quantified the role of functional rotation in the transport of a substrate, doxorubicin, by AcrB102,103. The functional rotation is the mechanism suggested by experimental studies as a possible process for the extrusion of a substrate19,104. By mimicking it, doxorubicin left the DBP and moved along the exit path102. The second work combined biased MD techniques to quantitatively evaluate the impact of the functional rotation on the transport of a substrate103. As result, the functional rotation reduced the free energy barrier associated with the efflux process and favoured the formation of a layer of structured waters, which crucially facilitated substrate transport by screening electrostatic interactions. Additionally, modelling more native conditions requires a focus on the sizes of the system to be modelled, and coarse-grained approaches have been developed to this goal105,106,107. For instance, Du et al.108 provided evidence that AcrZ109, a small protein interacting with AcrB, and the lipid environment synergistically activate an allosteric effect on the conformation of AcrB, affecting the substrate transport process, by combining experimental data with all-atoms and coarse-grained MD simulations. These examples demonstrate how computational simulations improve our comprehension of microscopic interactions, which are key to understanding the mechanism of drug resistance in clinical treatment.
Understanding the dialogue between the porin/transporter and the drug to guide drug design
Studying the dialogue occurring between the drug and its transporter is the key to determining the critical drug characteristics during the go/no-go steps. These include the involvement of globularity, flexibility, some moieties, and the distribution of charges along the molecules2,45,110,111,112,113,114.
In cellulo contributions to drug design
Accumulation assays are the method of choice for determining the structural parameters of compounds that favour their accumulation. LC-MS/MS measurements of accumulation allow the development of the notorious eNTRy rules, which stipulate that a compound displaying an ionisable nitrogen, low three-dimensionality and rigidity is most likely to accumulate in Gram-negative bacteria. These rules, together with the development of a complementary web tool (eNTRyway)50 and a detailed protocol44, provide the basis for the synthesis of new compounds targeting Gram-negative bacteria50,51. Recently, the Hergenrother group has improved these rules, identifying guanidinium as another positive functional group for uptake into E. coli46. Interestingly, the same group subsequently observed that these rules could not be applied to Pseudomonas aeruginosa due to its different membrane physiology47. They then developed the PASSage rules to characterize compounds with structures favourable for entry into P. aeruginosa cells and demonstrated that, unlike in E. coli, porins do not play a significant role in the entry of antibiotics into P. aeruginosa. Accumulation studies combined with fluorimetry measurements also allowed us to rank various fluoroquinolones according to their influx capacity and efflux susceptibility12. The influx ranking suggested the involvement of certain substituents as key factors for penetration.
In-silico contributions to drug design
The in-silico approaches described in the previous sections are essential for drug design, providing data on transport at microscopic resolution. For example, MD simulation is a powerful tool for visualising and dissecting possible pathways and constrictions. Well-designed single channel conductance measurements, in combination with MD simulations, are a highly effective method for investigating transport through individual components of the bacterial cell envelope at the molecular level8,9,115,116,117,118.
However, efficient drug design protocols require a high throughput that cannot be reached by several computational methods considered individually. A new approach has recently been developed to identify design rules for inhibitors of efflux systems. This approach makes use of a more statistical approach to docking poses, which allows interaction descriptors to be identified for machine learning approaches. Here, several configurations of the ligand extracted from MD simulations in water box119,120 or generated by conformation generators are docked to different configurations of the transporters. The residues most frequently in contact with the different poses of a ligand are interpreted as interaction descriptors72. These must be included in the set of data containing physico-chemical descriptors (LogP, volume, etc.), quantum chemistry descriptors (HOMO-LUMO gap, dipole, polarizability, etc.), and dynamical descriptors extracted from MD simulations of the ligands in the water box and inserted in the membrane (fluctuations, RMSD, minimum surface area, etc.) offer realistic data for machine learning approaches. The whole corpus of the descriptors mimics the fate of the compounds from structure to interaction with the bacterial transporters, making it ideal to approximate the different stages of compound-bacterium interactions.
Interestingly, several studies have succeeded in establishing clear patterns of characteristics that favors molecule entry into the bacteria cell. However, these studies often fail to consider the involvement of porins (presence or absence/type of porin/level of expression/mutation). Conversely, it has proved much more difficult to determine such patterns for efflux susceptibility because of the efflux pumps’ poly-specificity. Indeed, even slightly modified chemotypes follow different pathways through the bacterial membrane121. Likewise, fluoroquinolones are recognized and captured in a different way by the affinity sites (pocket) of AcrB, despite being structurally very similar12,30. This is a significant challenge given the crucial role of these transporters in the efficacy of the drug. Also of note is the observation that compounds with enhanced penetration tend to be efflux substrates12. Similarly, this poly-specificity presents a formidable challenge in the discovery of efflux pump inhibitors. Furthermore, the characteristics of influx and efflux vary considerably between species47,121, with this difference being amplified in clinical isolates.
Future perspective
The combination of different models has significantly improved our knowledge of how drugs are transported through bacterial membranes. These investigations have highlighted the complexity of drug transport, which depends on the environment, the fluidity of the membrane, the interactions between the transporter’s monomers/proteins, the charge of transporter residues, the charge and flexibility of the drug3,4,18. As Fig. 1 shows, significant gaps remain, and further efforts are needed to fill them.
Future research must address in vivo conditions as much as possible, paying close attention to the effects of time, drug concentration, ___location of drug target and environment influence. The next challenge is to determine the parameters of drug transport in situ, using an integrative analysis, which will allow us to provide keys in the clinical use of drugs to treat infections. Figure 3 illustrates how the new methods and tools can be used in an integrated combination to address key points and fill gaps (“A-E” in Fig. 1).
Determine transport kinetics closer to in vivo conditions
Various biases can occur during assays using so-called “reference” strains isolated several years ago that have very different characteristics due to their origin122,123, for example from the biological collections of the ATCC or the NCTC. Thus, the choice of these strains which may have different antibiotic susceptibilities between them and the comparison with recent clinical isolates can lead to misinterpretations. In addition, experiments sometimes use drug/cell concentrations that are not representative of therapeutic doses, and typically use standardized media that do not mimic the environment at the site of infection. Clearly, the transport of the same chemical can vary according to the species, the strain, and even from one cell to another. Furthermore, it also depends on cell density and community124,125 and, as illustrated in Fig. 3, on the environment of infectious sites, including nutrients, pH, ions, osmolarity, etc126., which impacts molecule ionizations, the energy available for active transport, transporter regulation127 and cell/membrane physiology59,128,129,130. Thus, it is essential that, where possible, the concentration of antibiotics in contact with the bacteria should be determined during clinical sampling. This will allow us to study the bacterial physiology and defense mechanisms more closely by confronting them at the same dose and under similar in situ conditions. This step aims to determine the RTC2T directly at the site of infection (Fig. 3).
The use of a combination of innovative tools such as LC/MS-MS and imaging will enable to monitor the in situ bacterial response with an integrated analysis of samplings (bacteria and antibiotic) (Fig. 3).
“The advancement of highly efficient equipment, particularly based on mass spectrometry, has simultaneously enhanced the potential of spatial biology131,132,133,134. Transposing the applications of spatial biology—focused on studying the in situ distribution of metabolites like peptides, proteins, and lipids, as well as pharmaceutical compounds133,135,136 —to investigate antibiotic distribution in infected tissues and its effects on metabolism would be highly beneficial. Currently, few studies mention the use of spatial metabolomics to study the metabolic state of pathogens within infectious sites137,138,139. With the recent development of sample preservation methods140, it would be interesting to extend this approach in the future to combat specific tissue infections (respiratory tract, digestive tract, etc.) requiring appropriate treatment.
At this moment, no reference of such application at clinical level has been reported, the benefit for the patient infected by resistant pathogens might stimulated the clinical assays. Consequently, we can envision using imaging techniques based on mass spectrometry to visualize the distribution of an antibiotic within a biofilm or infected tissue141,142,143, combined with the identification and characterization of the pathogen(s) phenotype, as well as the quantification of the antibiotic concentration in samples from the same tissue or biofilm. The combination of these techniques, previously applied in separate studies, will require technical adjustments. Moreover, these new methods and tools will provide key information to precisely quantify the role of degradative enzymes and target mutations in bacterial resistance, including their affinity for drugs and in situ kinetics (Fig. 1, Fig. 3). Currently, their involvement is only inferred from MIC measurements, which leaves a gap in our understanding of their respective in cellulo implications. This will make possible to understand the impact of ß-lactamase inhibitors or other inhibitors such as efflux pump inhibitors, on the internal antibiotic concentrations including diffusion rate and accumulation close to their respective target, and to develop a rational evaluation for future molecules.
Correlate the transport with real-time drug activity
Correlating RTC2T to the real-time inhibitory concentration (RTIC), is a major challenge because it includes the determination of the drug’s affinity for its target. We must also evaluate the intracellular drug concentration inducing a bacteriostatic/bactericidal effect. This will help define the concentration required in vivo to have an antibacterial action while reducing the resistance hazard. Additionally, unveiling the relationship between the total concentration and the RTIC in situ as a function of the pathogen species, its resistance profile, the conditions present in infectious site and its planktonic/biofilm organization, will specify the kinetic constants of drug transport in situ and finally establish the drug concentration to be used to induce an effective cell killing (Fig. 3, “T”).
Integrative view
Dynamic assessments of RT2CT-RTICs are essential for measuring the drug impact before resistant phenotypes emerge and alter the drug’s concentration at the target site. In a resistant strain, there is a race between achieving an appropriate RT2CT and the activation of resistance mechanisms such as the downregulation of porin expressions during the early stages of bacterial-antibiotic interaction3.
In this context, In vitro and in silico approaches play a pivotal role in pinpointing molecular and dynamic aspects. These methods (see Figs. 2 and 3) facilitate the refinement of discrete molecular interactions between molecule-transporter by using reconstituted systems in artificial membranes and enable the representation of the diffusion of the antibiotic within a 3D environment. For example, molecular modelling indicates the localisation and kinetics of mutual interactions and allows for the estimation of the energies occurring during the transport by considering the transporter and the molecule, key parameters for a future development of AI approach. Only by combining different disciplines such as biophysics, computational biology, microbiology, and others will we achieve a more comprehensive perspective on drug transport, and in turn, improve antibiotic therapies and the development of future drugs.
The membrane serves as the first line of defence in eucaryotic and procaryotic cells against toxic compounds, making it a critical focus in drug design. Drug design remains challenging because a drug must meet several criteria (good bioavailability and low toxicity, good permeability, propensity to be poorly shuttled out, non-cleavability by enzymes) to achieve the sufficient concentration on the target that ensure cell death and not induce the selection of new resistances.Therefore, it is essential to understand the bacterial strategy by using integrative, insightful and rational approaches including recent technologies and concepts6,144 (Fig. 3, “!A-!E”). Leveraging recent methodologies will facilitate the rational use of antibiotics best suited to the pathogen and its specific resistance profile.
References
European Centre for Disease Prevention and Control & World Health Organization. Antimicrobial Resistance Surveillance in Europe 2022 – 2020 Data. (World Health Organization. Regional Office for Europe, 2022).
Masi, M., Réfrégiers, M., Pos, K. M. & Pagès, J.-M. Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria. Nat. Microbiol. 2, 17001 (2017).
Vergalli, J. et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 18, 164–176 (2020).
Manrique, P. D., López, C. A., Gnanakaran, S., Rybenkov, V. V. & Zgurskaya, H. I. New understanding of multidrug efflux and permeation in antibiotic resistance, persistence, and heteroresistance. Ann. N. Y. Acad. Sci. 1519, 46–62 (2023).
Hegemann, J. D., Birkelbach, J., Walesch, S. & Müller, R. Current developments in antibiotic discovery. EMBO Rep. 24, e56184 (2023).
Sollier, J. et al. Revitalizing antibiotic discovery and development through in vitro modelling of in-patient conditions. Nat. Microbiol 9, 1–3 (2024).
Gervasoni, S. et al. Recognition of quinolone antibiotics by the multidrug efflux transporter MexB of Pseudomonas aeruginosa. Phys. Chem. Chem. Phys. 24, 16566–16575 (2022).
Acharya, A. et al. Conformational Dynamics of Loop L3 in OmpF: Implications toward Antibiotic Translocation and Voltage Gating. J. Chem. Inf. Model. 63, 910–927 (2023).
Acosta-Gutiérrez, S. et al. Getting drugs into Gram-negative bacteria: Rational rules for permeation through general porins. ACS Infect. Dis. 4, 1487–1498 (2018).
Acosta-Gutiérrez, S., Bodrenko, I. V. & Ceccarelli, M. The Influence of Permeability through Bacterial Porins in Whole-Cell Compound Accumulation. Antibiotics 10, 635 (2021).
Gervasoni, S. et al. Molecular determinants of avoidance and inhibition of Pseudomonas aeruginosa MexB efflux pump. mBio 14, e01403–e01423 (2023).
Vergalli, J. et al. The challenge of intracellular antibiotic accumulation, a function of fluoroquinolone influx versus bacterial efflux. Commun. Biol. 3, 1–12 (2020).
Dupont, M., James, C. E., Chevalier, J. & Pagès, J.-M. An early response to environmental stress involves regulation of OmpX and OmpF, two enterobacterial outer membrane pore-forming proteins. Antimicrob. Agents Chemother. 51, 3190–3198 (2007).
Pagès, J.-M., James, C. E. & Winterhalter, M. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 6, 893–903 (2008).
Bajaj, H. et al. Bacterial outer membrane porins as electrostatic nanosieves: Exploring transport rules of small polar molecules. ACS Nano 11, 5465–5473 (2017).
Scorciapino, M. A. et al. Rationalizing the permeation of polar antibiotics into Gram-negative bacteria. J. Phys.: Condens. Matter 29, 113001 (2017).
Davin-Regli, A., Pagès, J.-M. & Vergalli, J. The contribution of porins to enterobacterial drug resistance. J. Antimicrob. Chemother. 79, 2460–2470 (2024).
Du, D. et al. Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol. 16, 523–539 (2018).
Zgurskaya, H. I., Walker, J. K., Parks, J. M. & Rybenkov, V. V. Multidrug efflux pumps and the two-faced janus of substrates and inhibitors. Acc. Chem. Res. 54, 930–939 (2021).
Westfall, D. A. et al. Bifurcation kinetics of drug uptake by Gram-negative bacteria. PLoS ONE 12, e0184671 (2017).
Rybenkov, V. V. et al. The Whole Is Bigger than the Sum of Its Parts: Drug Transport in the Context of Two Membranes with Active Efflux. Chem. Rev. 121, 5597–5631 (2021).
Saha, P., Sikdar, S., Krishnamoorthy, G., Zgurskaya, H. I. & Rybenkov, V. V. Drug Permeation against Efflux by Two Transporters. ACS Infect. Dis. 6, 747–758 (2020).
Krishnamoorthy, G. et al. Synergy between active efflux and outer membrane diffusion defines rules of antibiotic permeation into Gram-negative bacteria. mBio 8, e01172-17 (2017).
Pinet, E. et al. A simple phenotypic test for detecting the contribution of outer membrane permeability to carbapenem resistance. J. Med. Microbiol. 69, 63–71 (2020).
Castanheira, M., Doyle, T. B., Hubler, C., Sader, H. S. & Mendes, R. E. Ceftazidime-avibactam activity against a challenge set of carbapenem-resistant Enterobacterales: Ompk36 L3 alterations and β-lactamases with ceftazidime hydrolytic activity lead to elevated MIC values. Int J. Antimicrob. Agents 56, 106011 (2020).
Al-Marzooq, F., Ghazawi, A., Tariq, S., Daoud, L. & Collyns, T. Discerning the role of polymyxin B nonapeptide in restoring the antibacterial activity of azithromycin against antibiotic-resistant Escherichia coli. Front Microbiol 13, 998671 (2022).
Ferrand, A. et al. Contribution of efflux and mutations in fluoroquinolone susceptibility in MDR enterobacterial isolates: a quantitative and molecular study. J. Antimicrob. Chemother. 78, 1532–1542 (2023).
Lamers, R. P., Cavallari, J. F. & Burrows, L. L. The Efflux Inhibitor Phenylalanine-Arginine Beta-Naphthylamide (PAβN) Permeabilizes the Outer Membrane of Gram-Negative Bacteria. PLoS One 8, e60666 (2013).
Schuster, S., Bohnert, J. A., Vavra, M., Rossen, J. W. & Kern, W. V. Proof of an Outer Membrane Target of the Efflux Inhibitor Phe-Arg-β-Naphthylamide from Random Mutagenesis. Molecules 24, 470 (2019).
Vergalli, J. et al. A framework for dissecting affinities of multidrug efflux transporter AcrB to fluoroquinolones. Commun. Biol. 5, 1–11 (2022).
Camp, J. et al. Limited Multidrug Resistance Efflux Pump Overexpression among Multidrug-Resistant Escherichia coli Strains of ST131. Antimicrob. Agents Chemother. 65, e01735-20 (2021).
Blair, J. M. A. & Piddock, L. J. V. How to measure export via bacterial multidrug resistance efflux pumps. mBio 7, e00840–16 (2016).
Masi, M. et al. Fluorescence enlightens RND pump activity and the intrabacterial concentration of antibiotics. Res. Microbiol. 169, 432–441 (2018).
Masi, M. et al. Cephalosporin translocation across enterobacterial OmpF and OmpC channels, a filter across the outer membrane. Commun. Biol. 5, 1–10 (2022).
Vidal-Aroca, F., Meng, A., Minz, T., Page, M. G. P. & Dreier, J. Use of resazurin to detect mefloquine as an efflux-pump inhibitor in Pseudomonas aeruginosa and Escherichia coli. J. Microbiological Methods 79, 232–237 (2009).
Stone, M. R. L. et al. Fluoroquinolone-derived fluorescent probes for studies of bacterial penetration and efflux. Med. Chem. Commun. https://doi.org/10.1039/C9MD00124G (2019).
Stone, M. R. L. et al. Fluorescent macrolide probes – synthesis and use in evaluation of bacterial resistance. RSC Chem. Biol. https://doi.org/10.1039/D0CB00118J (2020).
Whittle, E. E. et al. Efflux Impacts Intracellular Accumulation Only in Actively Growing Bacterial Cells. mBio 12, e02608-21 (2021).
Zhang, B. et al. Synthesis of vancomycin fluorescent probes that retain antimicrobial activity, identify Gram-positive bacteria, and detect Gram-negative outer membrane damage. Commun. Biol. 6, 1–16 (2023).
Whittle, E. E. et al. Flow Cytometric Analysis of Efflux by Dye Accumulation. Front. Microbiol. 10, 2319 (2019).
Cai, H., Rose, K., Liang, L.-H., Dunham, S. & Stover, C. Development of a liquid chromatography/mass spectrometry-based drug accumulation assay in Pseudomonas aeruginosa. Anal. Biochem. 385, 321–325 (2009).
Dumont, E. et al. Antibiotics and efflux: combined spectrofluorimetry and mass spectrometry to evaluate the involvement of concentration and efflux activity in antibiotic intracellular accumulation. J. Antimicrob. Chemother. 74, 58–65 (2019).
Vergalli, J. et al. Spectrofluorimetric quantification of antibiotic drug concentration in bacterial cells for the characterization of translocation across bacterial membranes. Nat. Protoc. 13, 1348–1361 (2018).
Geddes, E. J., Li, Z. & Hergenrother, P. J. An LC-MS/MS assay and complementary web-based tool to quantify and predict compound accumulation in E. coli. Nat. Protoc. 16, 4833–4854 (2021).
Richter, M. F. et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017).
Perlmutter, S. J. et al. Compound Uptake into E. coli Can Be Facilitated by N-Alkyl Guanidiniums and Pyridiniums. ACS Infect. Dis. 7, 162–173 (2021).
Cain, B. N. & Hergenrother, P. J. Using permeation guidelines to design new antibiotics—A PASsagE into Pseudomonas aeruginosa. Clin. Transl. Med 14, e1600 (2024).
Geddes, E. J. et al. Porin-independent accumulation in Pseudomonas enables antibiotic discovery. Nature 624, 145–153 (2023).
Richter, M. F. & Hergenrother, P. J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci. 1435, 18–38 (2019).
Parker, E. N. et al. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol 5, 67–75 (2020).
Muñoz, K. A. & Hergenrother, P. J. Facilitating Compound Entry as a Means to Discover Antibiotics for Gram-Negative Bacteria. Acc. Chem. Res 54, 1322–1333 (2021).
Kaščáková, S., Maigre, L., Chevalier, J., Réfrégiers, M. & Pagès, J.-M. Antibiotic transport in resistant bacteria: Synchrotron UV fluorescence microscopy to determine antibiotic accumulation with single cell resolution. PLoS One 7, e38624 (2012).
Rosas, N. C. et al. The evolutionary mechanism of non-carbapenemase carbapenem-resistant phenotypes in Klebsiella spp. eLife 12, e83107 (2023).
Ma, Y., Ramoneda, J. & Johnson, D. R. Timing of antibiotic administration determines the spread of plasmid-encoded antibiotic resistance during microbial range expansion. Nat. Commun. 14, 3530 (2023).
Vergalli, J. et al. Fluoroquinolone structure and translocation flux across bacterial membrane. Sci. Rep. 7, 9821 (2017).
Łapińska, U. et al. Fast bacterial growth reduces antibiotic accumulation and efficacy. eLife 11, e74062 (2022).
Schrader, S. M., Botella, H. & Vaubourgeix, J. Reframing antimicrobial resistance as a continuous spectrum of manifestations. Curr. Opin. Microbiol. 72, 102259 (2023).
Levin-Reisman, I. et al. Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017).
Paracini, N., Schneck, E., Imberty, A. & Micciulla, S. Lipopolysaccharides at Solid and Liquid Interfaces: Models for Biophysical Studies of the Gram-negative Bacterial Outer Membrane. Adv. Colloid Interface Sci. 301, 102603 (2022).
Prajapati, J. D., Kleinekathöfer, U. & Winterhalter, M. How to Enter a Bacterium: Bacterial Porins and the Permeation of Antibiotics. Chem. Rev. 121, 5158–5192 (2021).
Kojima, S. & Nikaido, H. Permeation rates of penicillins indicate that Escherichia coli porins function principally as nonspecific channels. PNAS 110, E2629–E2634 (2013).
Ghai, I. et al. General method to determine the flux of charged molecules through nanopores applied to β-lactamase inhibitors and OmpF. J. Phys. Chem. Lett. 8, 1295–1301 (2017).
Biedermann, F., Ghale, G., Hennig, A. & Nau, W. M. Fluorescent artificial receptor-based membrane assay (FARMA) for spatiotemporally resolved monitoring of biomembrane permeability. Commun. Biol. 3, 383 (2020).
Terrasse, R. & Winterhalter, M. Translocation of small molecules through engineered outer-membrane channels from Gram-negative bacteria. Eur. Phys. J. E 41, 111 (2018).
Wang, J., Terrasse, R., Bafna, J. A., Benier, L. & Winterhalter, M. Electrophysiological Characterization of Transport Across Outer‐Membrane Channels from Gram‐Negative Bacteria in Presence of Lipopolysaccharides. Angew. Chem. Int Ed. Engl. 59, 8517–8521 (2020).
Bafna, J. A. et al. Kanamycin uptake into Escherichia coli is facilitated by OmpF and OmpC porin channels located in the outer membrane. ACS Infect. Dis. 6, 1855–1865 (2020).
Zuo, K. et al. Metadynamics simulations of ligands binding to protein surfaces: a novel tool for rational drug design. Phys. Chem. Chem. Phys. 25, 13819–13824 (2023).
Manathunga, M., Götz, A. W. & Merz, K. M. Computer-aided drug design, quantum-mechanical methods for biological problems. Curr. Opin. Struct. Biol. 75, 102417 (2022).
Wang, Y. et al. Retro Drug Design: From Target Properties to Molecular Structures. J. Chem. Inf. Model. 62, 2659–2669 (2022).
Gorgulla, C., Jayaraj, A., Fackeldey, K. & Arthanari, H. Emerging Frontiers in Virtual Drug Discovery: From Quantum Mechanical Methods to Deep Learning Approaches. Curr. Opin. Chem. Biol. 69, 102156 (2022).
Wong, F., de la Fuente-Nunez, C. & Collins, J. J. Leveraging artificial intelligence in the fight against infectious diseases. Science 381, 164–170 (2023).
Mehla, J. et al. Predictive Rules of Efflux Inhibition and Avoidance in Pseudomonas aeruginosa. mBio 12, e02785–20 (2021).
Carugo, O. & Djinović-Carugo, K. Structural biology: A golden era. PLOS Biol. 21, e3002187 (2023).
Nitta, R., Imasaki, T. & Nitta, E. Recent progress in structural biology: lessons from our research history. Microscopy 67, 187–195 (2018).
Du, Z. et al. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 16, 5634–5651 (2021).
Graille, M., Sacquin-Mora, S. & Taly, A. Best Practices of Using AI-Based Models in Crystallography and Their Impact in Structural Biology. J. Chem. Inf. Model. 63, 3637–3646 (2023).
Jisna, V. A. & Jayaraj, P. B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J. 40, 522–544 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a 3-track neural network. Science 373, 871–876 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493 (2024).
Subramaniam, S. & Kleywegt, G. J. A paradigm shift in structural biology. Nat. Methods 19, 20–23 (2022).
Oliva, F. et al. Modelling eNvironment for Isoforms (MoNvIso): A general platform to predict structural determinants of protein isoforms in genetic diseases. Front Chem. 10, 1059593 (2023).
Shen, M. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006).
Ramaswamy, V. K., Vargiu, A. V., Malloci, G., Dreier, J. & Ruggerone, P. Molecular Rationale behind the Differential Substrate Specificity of Bacterial RND Multi-Drug Transporters. Sci. Rep. 7, 8075 (2017).
Ramaswamy, V. K., Vargiu, A. V., Malloci, G., Dreier, J. & Ruggerone, P. Molecular Determinants of the Promiscuity of MexB and MexY Multidrug Transporters of Pseudomonas aeruginosa. Front Microbiol 9, 1144 (2018).
Catte, A. et al. Common recognition topology of mex transporters of Pseudomonas aeruginosa revealed by molecular modelling. Front Pharm. 13, 1021916 (2022).
Halperin, I., Ma, B., Wolfson, H. & Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins: Struct., Funct., Bioinforma. 47, 409–443 (2002).
Brooijmans, N. & Kuntz, I. D. Molecular recognition and docking algorithms. Annu Rev. Biophys. Biomol. Struct. 32, 335–373 (2003).
Gentile, F., Oprea, T. I., Tropsha, A. & Cherkasov, A. Surely you are joking, Mr Docking! Chem. Soc. Rev. 52, 872–878 (2023).
Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 36, 78–95 (2015).
Tam, H.-K. et al. Binding and Transport of Carboxylated Drugs by the Multidrug Transporter AcrB. J. Mol. Biol. 432, 861–877 (2020).
Atzori, A. et al. Molecular Interactions of Cephalosporins with the deep binding pocket of the RND Transporter AcrB. J. Phys. Chem. B 123, 4625–4635 (2019).
Zhang, B., Li, H., Yu, K. & Jin, Z. Molecular docking-based computational platform for high-throughput virtual screening. CCF Trans. High. Perform. Comput 4, 63–74 (2022).
Sinha, S., Tam, B. & Wang, S. M. Applications of Molecular Dynamics Simulation in Protein Study. Membr. (Basel) 12, 844 (2022).
Raghavan, B. et al. Drug Design in the Exascale Era: A Perspective from Massively Parallel QM/MM Simulations. J. Chem. Inf. Model 63, 3647–3658 (2023).
Wolf, S. Predicting Protein–Ligand Binding and Unbinding Kinetics with Biased MD Simulations and Coarse-Graining of Dynamics: Current State and Challenges. J. Chem. Inf. Model. 63, 2902–2910 (2023).
Bock, L. V., Gabrielli, S., Kolář, M. H. & Grubmüller, H. Simulation of Complex Biomolecular Systems: The Ribosome Challenge. Annu Rev. Biophys. 52, 361–390 (2023).
Chen, H. & Chipot, C. Enhancing sampling with free-energy calculations. Curr. Opin. Struct. Biol. 77, 102497 (2022).
Kamenik, A. S., Linker, S. M. & Riniker, S. Enhanced sampling without borders: on global biasing functions and how to reweight them. Phys. Chem. Chem. Phys. 24, 1225–1236 (2022).
Ray, D., Ansari, N., Rizzi, V., Invernizzi, M. & Parrinello, M. Rare Event Kinetics from Adaptive Bias Enhanced Sampling. J. Chem. Theory Comput 18, 6500–6509 (2022).
Biarnés, X., Bongarzone, S., Vargiu, A. V., Carloni, P. & Ruggerone, P. Molecular motions in drug design: the coming age of the metadynamics method. J. Comput Aided Mol. Des. 25, 395–402 (2011).
Schulz, R., Vargiu, A. V., Collu, F., Kleinekathöfer, U. & Ruggerone, P. Functional Rotation of the Transporter AcrB: Insights into Drug Extrusion from Simulations. PLoS Comput Biol. 6, e1000806 (2010).
Vargiu, A. V. et al. Water-mediated interactions enable smooth substrate transport in a bacterial efflux pump. Biochim. Biophys. Acta Gen. Subj. 1862, 836–845 (2018).
Alav, I. et al. Structure, assembly, and function of tripartite efflux and type 1 secretion systems in Gram-negative bacteria. Chem. Rev. 121, 5479–5596 (2021).
Jin, J., Pak, A. J., Durumeric, A. E. P., Loose, T. D. & Voth, G. A. Bottom-up Coarse-Graining: Principles and Perspectives. J. Chem. Theory Comput 18, 5759–5791 (2022).
Jefferies, D. & Khalid, S. Atomistic and coarse-grained simulations of membrane proteins: A practical guide. Methods 185, 15–27 (2021).
Marrink, S. J. et al. Two decades of Martini: Better beads, broader scope. WIREs Computational Mol. Sci. 13, e1620 (2023).
Du, D. et al. Interactions of a Bacterial RND Transporter with a Transmembrane Small Protein in a Lipid Environment. Structure 28, 625–634.e6 (2020).
Hobbs, E. C., Yin, X., Paul, B. J., Astarita, J. L. & Storz, G. Conserved small protein associates with the multidrug efflux pump AcrB and differentially affects antibiotic resistance. Proc. Natl Acad. Sci. 109, 16696–16701 (2012).
Nikaido, H. & Rosenberg, E. Y. Effect on solute size on diffusion rates through the transmembrane pores of the outer membrane of Escherichia coli. J. Gen. Physiol. 77, 121–135 (1981).
Brown, D. G., May-Dracka, T. L., Gagnon, M. M. & Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 57, 10144–10161 (2014).
Nakae, T. & Nikaido, H. Outer membrane as a diffusion barrier in Salmonella typhimurium. Penetration of oligo- and polysaccharides into isolated outer membrane vesicles and cells with degraded peptidoglycan layer. J. Biol. Chem. 250, 7359–7365 (1975).
Ruggiu, F. et al. Size Matters and How You Measure It: A Gram-Negative Antibacterial Example Exceeding Typical Molecular Weight Limits. ACS Infect. Dis. 5, 1688–1692 (2019).
O’Shea, R. & Moser, H. E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 51, 2871–2878 (2008).
Milenkovic, S. et al. How the physical properties of bacterial porins match environmental conditions. Phys. Chem. Chem. Phys. 25, 12712–12722 (2023).
Acharya, A., Prajapati, J. D. & Kleinekathöfer, U. Atomistic Simulation of Molecules Interacting with Biological Nanopores: From Current Understanding to Future Directions. J. Phys. Chem. B 126, 3995–4008 (2022).
Vasan, A. K. et al. Role of internal loop dynamics in antibiotic permeability of outer membrane porins. Proc. Natl Acad. Sci. 119, e2117009119 (2022).
Acharya, A., Jana, K., Gurvic, D., Zachariae, U. & Kleinekathöfer, U. Fast prediction of antibiotic permeability through membrane channels using Brownian dynamics. Biophysical J. 122, 2996–3007 (2023).
Malloci, G. et al. A database of force-field parameters, dynamics, and properties of antimicrobial compounds. Molecules 20, 13997–14021 (2015).
Gervasoni, S. et al. AB-DB: Force-Field parameters, MD trajectories, QM-based data, and Descriptors of Antimicrobials. Sci. Data 9, 148 (2022).
Leus, I. V. et al. Functional Diversity of Gram-Negative Permeability Barriers Reflected in Antibacterial Activities and Intracellular Accumulation of Antibiotics. Antimicrobial Agents Chemother. 67, e01377-22 (2023).
Cendra, M. D. M. & Torrents, E. Differential adaptability between reference strains and clinical isolates of Pseudomonas aeruginosa into the lung epithelium intracellular lifestyle. Virulence 11, 862 (2020).
Siroy, A. et al. Global comparison of the membrane subproteomes between a multidrug-resistant Acinetobacter baumannii strain and a reference strain. J. Proteome Res 5, 3385–3398 (2006).
Zhang, Q. et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science 333, 1764–1767 (2011).
Wen, X., Langevin, A. M. & Dunlop, M. J. Antibiotic export by efflux pumps affects growth of neighboring bacteria. Sci. Rep. 8, 15120 (2018).
Littlejohn, P. T. et al. Multiple micronutrient deficiencies in early life cause multi-kingdom alterations in the gut microbiome and intrinsic antibiotic resistance genes in mice. Nat. Microbiol 8, 2392–2405 (2023).
Ferrand, A., Vergalli, J., Pagès, J.-M. & Davin-Regli, A. An intertwined network of regulation controls membrane permeability including drug influx and efflux in Enterobacteriaceae. Microorganisms 8, 833 (2020).
Bergmiller, T. et al. Biased partitioning of the multidrug efflux pump AcrAB-TolC underlies long-lived phenotypic heterogeneity. Science 356, 311–315 (2017).
Evans, K. & Poole, K. The MexA-MexB-OprM multidrug efflux system of Pseudomonas aeruginosa is growth-phase regulated. FEMS Microbiol. Lett. 173, 35–39 (1999).
Thorfinnsdottir, L. B., Bø, G. H., Booth, J. A. & Bruheim, P. Survival of Escherichia coli after high-antibiotic stress is dependent on both the pregrown physiological state and incubation conditions. Front. Microbiol. 14, 1149978 (2023).
Buck, A. & Walch, A. In Situ Drug and Metabolite Analysis in Biological and Clinical Research by MALDI MS Imaging. Bioanalysis 6, 1241–1253 (2014).
Jia, F., Zhao, X. & Zhao, Y. Advancements in ToF-SIMS imaging for life sciences. Front. Chem. 11, 1237408 (2023).
Chen, Y. et al. Recent Advances in Mass Spectrometry-Based Spatially Resolved Molecular Imaging of Drug Disposition and Metabolomics. Drug Metab. Dispos. 51, 1273–1283 (2023).
Xiao, Y. et al. Recent advances of ambient mass spectrometry imaging for biological tissues: A review. Anal. Chim. Acta 1117, 74–88 (2020).
Giordano, S. et al. 3D Mass Spectrometry Imaging Reveals a Very Heterogeneous Drug Distribution in Tumors. Sci. Rep. 6, 37027 (2016).
Zhang, J. et al. Evaluation of the tumor-targeting efficiency and intratumor heterogeneity of anticancer drugs using quantitative mass spectrometry imaging. Theranostics 10, 2621 (2020).
Dean, D. A. et al. Spatial Metabolomics Reveals Localized Impact of Influenza Virus Infection on the Lung Tissue Metabolome. mSystems 7, e00353 (2022).
Aksenov, A. A. et al. Spatial chemistry of citrus reveals molecules bactericidal to Candidatus Liberibacter asiaticus. Sci. Rep. 14, 20306 (2024).
Man, L., Klare, W. P., Dale, A. L., Cain, J. A. & Cordwell, S. J. Integrated mass spectrometry-based multi-omics for elucidating mechanisms of bacterial virulence. Biochemical Soc. Trans. 49, 1905–1926 (2021).
Dannhorn, A. et al. Morphological and molecular preservation through universal preparation of fresh-frozen tissue samples for multimodal imaging workflows. Nat. Protoc. 19, 2685–2711 (2024).
McCaughey, C., Trebino, M., Yildiz, F. H. & Sanchez, L. M. Utilizing Imaging Mass Spectrometry to Analyze Microbial Biofilm Chemical Responses to Exogenous Compounds. Methods Enzymol. 665, 281–304 (2022).
Parmar, D., Rosado-Rosa, J. M., Shrout, J. D. & Sweedler, J. V. Metabolic insights from mass spectrometry imaging of biofilms: A perspective from model microorganisms. Methods 224, 21–34 (2024).
Cheng, S.-H. et al. Multimodal imaging distribution assessment of a liposomal antibiotic in an infectious disease model. J. Controlled Release 352, 199–210 (2022).
Wong, F. et al. Discovery of a structural class of antibiotics with explainable deep learning. Nature 626, 177–185 (2024).
Paixão, L. et al. Fluorometric determination of ethidium bromide efflux kinetics in Escherichia coli. J. Biol. Eng. 3, 18 (2009).
Borselli, D., Brunel, J. M., Gorgé, O. & Bolla, J. M. Polyamino-Isoprenyl Derivatives as Antibiotic Adjuvants and Motility Inhibitors for Bordetella bronchiseptica Porcine Pulmonary Infection Treatment. Front Microbiol 10, 1771 (2019).
Nagano, K. & Nikaido, H. Kinetic behavior of the major multidrug efflux pump AcrB of Escherichia coli. Proc. Natl Acad. Sci. USA 106, 5854–5858 (2009).
Haynes, M. K. et al. High-Throughput Flow Cytometry Screening of Multidrug Efflux Systems. in Bacterial Multidrug Exporters: Methods and Protocols (eds. Yamaguchi, A. & Nishino, K.) 293–318 (Springer, New York, NY, 2018).
Malléa, M., Chevalier, J., Eyraud, A. & Pagès, J.-M. Inhibitors of antibiotic efflux pump in resistant Enterobacter aerogenes strains. Biochemical Biophysical Res. Commun. 293, 1370–1373 (2002).
Wang-Kan, X. et al. Lack of AcrB Efflux Function Confers Loss of Virulence on Salmonella enterica Serovar Typhimurium. mBio 8, e00968-17 (2017).
Cama, J. et al. Single-cell microfluidics facilitates the rapid quantification of antibiotic accumulation in Gram-negative bacteria. Lab a Chip 20, 2765–2775 (2020).
Prochnow, H. et al. Subcellular Quantification of Uptake in Gram-Negative Bacteria. Anal. Chem. 91, 1863–1872 (2019).
Tian, H., Six, D. A., Krucker, T., Leeds, J. A. & Winograd, N. Subcellular chemical imaging of antibiotics in single bacteria using C60-secondary ion mass spectrometry. Anal. Chem. 89, 5050–5057 (2017).
Vargiu, A. V. et al. Computer simulations of the activity of RND efflux pumps. Res. Microbiol. 169, 384–392 (2018).
Acknowledgements
We thank, A. Davin-Regli, JM. Bolla, JM. Brunel and M. Masi for their fruitful discussions. This work was supported by Aix-Marseille Univ., INSERM, Service de Santé des Armées, by Soleil program, by the Health Extended ALliance for Innovative Therapies, Advanced Lab-research, and Integrated Approaches of Precision Medicine partnership (HEAL ITALIA), foundation by the Italian Ministry of University and Research, PNRR, mission 4, component 2, investment 1.3, project number PE00000019 (University of Cagliari) and under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.5 - Call for tender No.3277 published on December 30, 2021 by the Italian Ministry of University and Research (MUR) funded by the European Union – NextGenerationEU. Project Code ECS0000038 – Project Title eINS Ecosystem of Innovation for Next Generation Sardinia – CUP J85B17000360007 - Concession Decree No. 1056 adopted on June 23, 2022 by the Italian Ministry of Ministry of University and Research (MUR).
Author information
Authors and Affiliations
Contributions
J.V., M.R., P.R., M.W., J-M.P.: writing, review, and final editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Ranjana Pathania and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vergalli, J., Réfrégiers, M., Ruggerone, P. et al. Advances in methods and concepts provide new insight into antibiotic fluxes across the bacterial membrane. Commun Biol 7, 1508 (2024). https://doi.org/10.1038/s42003-024-07168-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-07168-4
