
Abstract
Background
Early detection of Parkinson’s disease (PD), a neurodegenerative disease with central and peripheral nerve involvement, ensures timely treatment access. Microbes influence nervous system health and are altered in PD.
Methods
We examined gut and mouth microbiomes from recently diagnosed patients in a geographically diverse, matched case-control, shotgun metagenomics study.
Results
Here, we show greater alpha-diversity in 445 PD patients versus 221 controls. The microbial signature of PD includes overabundance of 16 OTUs, including Streptococcus mutans and Bifidobacterium dentium, and depletion of 28 OTUs. Machine learning models indicate that subspecies level oral microbiome abundances best distinguish PD with reasonably high accuracy (area under the curve: 0.758). Microbial networks are disrupted in cases, with reduced connectivity between short-chain fatty acid-producing bacteria the the gut. Importantly, microbiome diversity metrics are associated with non-motor autonomic symptom severity.
Conclusions
Our results provide evidence that predictive oral PD microbiome signatures could possibly be used as biomarkers for the early detection of PD, particularly when there is peripheral nervous system involvement.
Plain language summary
Parkinson’s disease (PD) is a neurodegenerative disease that is characterized by both motor symptoms, such as tremors, and non-motor symptoms, such as constipation. Our aim was to determine whether there were differences in the number and types of microbes living in the saliva and intestines of people with and without PD. We saw significant differences in the microbial communities living in healthy controls compared to people with PD. Additionally, we found that the proportions of microbe types in saliva were the best at distinguishing between controls and cases, and identified the specific kinds of microbes that were driving this distinction. These results highlight the potential importance of the saliva microbiome in understanding the causes and symptomatology of PD.
Similar content being viewed by others

Introduction
The gut is a densely innervated organ controlled by its own enteric nervous system with oversight from the autonomic nervous system and brain. The microbiota-gut-brain axis represents a bidirectional connection between the gastrointestinal system, and the microbes that inhabit it, with the central nervous system (CNS)1,2,3.
In early Parkinson’s disease (PD), the microbial community of the gut is altered before motor symptoms fully manifest4,5,6, and may play a role in disease susceptibility6,7. At autopsy, ɑ-synuclein-containing Lewy bodies, which are the pathological hallmark of PD8, are found at all levels throughout the microbiota-gut-brain axis, including the central, autonomic, and enteric neurons9. It is thought that alterations in the gut microbial community increase intestinal permeability9, cause immune cell activation10, and initiate ɑ-synuclein aggregation11. The gut may then become a source of seeding ɑ-synuclein12 which is trafficked to the brain. Lewy bodies containing ɑ-synuclein are detectable throughout the gastrointestinal tract13,14, including in the salivary glands of both prodromal and clinically manifest PD cases15 This suggests the possibility of pathological ɑ-synuclein spread from the mouth and nose to the brain as one potential route of protein seeding in the early stages of the disease process. This potential etiology provides rationale for examining the oral microbiome to understand whether microorganisms at this ___location play a role. Microbiome perturbation also appears to be important in the later stages of PD, as taxonomic shifts delay the absorption of dopaminergic drugs16 and correlate with more severe clinical symptoms including PD dementia17,18,19.
Most previous studies of the PD microbiome have been relatively small (fewer than 200 participants per cohort), geographically restricted, utilized variable techniques, and primarily focused on the analysis of stool samples4,20,21,22,23,24,25,26,27,28,29,30. Despite these limitations, specific intestinal microbe genera are consistently reported as overabundant in PD such as Akkermansia, Alistipes, Bifidobacterium, and Streptococcus, while others including Coprococcus, Faecalibacterium, Prevotella, and Roseburia are depleted9. The microbial diversity of the gut appears to be increased in patients with PD9, suggesting host control over some taxa is diminished. At a functional level, the stool of patients with PD shows depletion in bacteria involved in short-chain fatty acid synthesis10,11,21,31, glutamate metabolism32,33,34, and increased abundance of bacteria capable of amino acid degradation33,34. While much less is known about the PD oral microbiome, there is evidence that it too is perturbed15,35,36,37,38. Previous studies demonstrated significant differences in taxonomic and functional compositions of saliva microbiomes between PD cases and healthy controls36,37,39.
If distinct microbial changes occur throughout the gastrointestinal system during PD onset and progression, we hypothesize that these changes are related to disease burden (i.e., symptoms). Using a large, geographically diverse, matched case-control design, we aimed to determine (1) whether previously reported associations between PD and the oral and intestinal microbiomes were observed using shallow shotgun metagenomic sequencing; (2) whether microbiome alterations in PD correlated with the severity of clinical symptoms; (3) whether disease-relevant covariates better explained differential relative abundances of taxa and functions relative to PD diagnosis; and (4) whether oral microbiome sampling, which is easier to obtain than gut samples, could statistically predict PD cases and controls.
In this study, we investigate differences in the diversity and composition of both stool and saliva microbiomes between PD cases and healthy controls. We also identify taxa that discriminate between these two populations, and interrogate disruption in the microbial co-abundance networks within the PD gut microbiome. Lastly, we compare the ability of saliva and the stool microbiome to distinguish between PD cases and controls, and find that the abundances of saliva taxa provide the best information for making this distinction. This work highlights the potential importance of not just the gut microbiome, but also the oral microbiome in understanding the etiology and progression of PD.
Methods
Study design
See Supplementary Fig. 1 for a graphical depiction of the study design and methods. We recruited 774 (304 controls and 470 cases) participants for a matched case-control study, through 23andMe’s online Research Program. The inclusion criteria for participants with PD were: (1) a self-reported PD diagnosis; (2) disease duration <5 years (amended to <8 years to meet enrollment goals); and (3) current age between 50 and 69 years. Age and sex matched controls who did not report a diagnosis of PD nor family history of PD were randomly selected from 23andMe’s Research cohort. The exclusion criteria for both groups included: (1) positive LRRK2 G2019S or GBA N370S carrier status; (2) antibiotic usage within the last 6 months; (3) self-reported vegetarian/vegan diet; (4) non-European genetic ancestry; and (5) mailing addresses outside the US. The study was approved by Salus IRB as an observational study (protocol No. US-IRB-26, AAHRPP-accredited IRB), and all participants gave informed consent. To enable successful matching, cases were recruited first, and, once the enrollment target was met, the mean age and sex profile of the PD cohort was established. Controls were then enrolled by a rolling manual recruitment process, to match age ranges and sex profile of the PD cases. The total participants on which the analyses were performed was reduced due to sequencing quality control (QC), missing age data or age outside of inclusion criteria (controls only), removing participants who had experienced gastroenteritis (after noting that their stool microbiome composition was quite different from all others), and after rarefaction of reads (in which we remove samples with total read counts fewer than 10,000). The sample sizes after each step that reduced sample size can be seen in Supplementary Fig. 2.
After signing consent, barcoded stool and saliva collection kits with standardized collection instructions were shipped to participants (OMNIgene•GUT and OMNIgene•ORAL; DNA Genotek, Ontario, Canada). Participants were instructed to refrain from eating, drinking, chewing gum, smoking, or brushing teeth 30 min before saliva collection. Kits contained reagents to rapidly homogenize samples at the point of collection and maintain the DNA stable at ambient temperature for 60 days, with no cold chain required. Participants were instructed to return their samples in prepaid mailing boxes within 2 weeks of collection.
Phenotypic data collection
After collecting and returning their samples, participants were instructed to complete a 119 item online survey that included: (1) demographics; (2) biometrics; (3) medication usage (including laxatives/stool softeners and anti-Parkinsonian medications in cases only); (4) symptoms of PD disease (cases only); (5) diet, caffeine, and alcohol consumption; (6) dental cleaning habits; (7) and Bristol stool scores40 with illustrative examples of consistency. An additional 102 survey answers were collected from patients with PD to further characterize their clinical phenotype41.
Metagenomic sequencing and processing
DNA extraction, sequencing, and annotation were performed by Diversigen (Minnesota, USA). Visual inspections of the collection kits as they arrived were conducted and recorded, including sample discoloration, low volume, and incorrect usage. Samples were randomized across nine batches, with all batches containing both case and control samples. Additional QC included DNA quantification to determine whether there were at least 16,000 16S copies/μL and greater than 1 ng/µL of DNA per sample. Microbial DNA extraction was performed at room temperature from saliva and stool samples using the QIAGEN Powersoil Pro DNA isolation kit with a liquid-handling robot, according to kit protocols (QIAGEN, Netherlands). Sequence libraries were prepared using enzymatic tagmentation with low cycle polymerase chain reaction. 88 samples plus a positive and negative library preparation control were included in each 96 well-plate. Sequencing was conducted on the BoosterShot® shallow shotgun sequencing platform, with 1 × 100 reads at a target depth of 2 million reads per sample. The Cutadapt42 software tool was used to discard sequences with a mean quality score less than 30. Samples with fewer than 10,000 reads were discarded. The remaining quality reads were annotated taxonomically by alignment to a database containing all representative genomes in NCBI’s RefSeq for bacteria with additional manually curated strains (Diversigen). Alignments were made at 97% identity against all reference genomes. Each input sequence was compared to every reference sequence in the Diversigen Venti database using fully gapped alignment with BURST (Al-Ghalith & Knights 2020), and ties were broken through minimization of the total number of unique Operational Taxonomic Units (OTUs). Each input sequence was assigned the lowest common ancestor that was consistent across at least 80% of all reference sequences tied for best hit. For functional group annotation, Kyoto Encyclopedia of Genes and Genomes Orthology groups (KEGG) orthologies (KOs) were assigned to reads via the Prokka software tool43, at 97% identity against a gene database derived from the same strain database used for taxonomic annotation (Venti). KOs were collapsed to higher levels as well: level-2 and −3 KEGG pathways and KEGG Modules. To avoid incorporation of spurious microbiome features into the analyses, OTUs and KOs with total relative abundances <1e-4 or prevalent in <10% of participants were discarded. Additionally we removed taxa that could not be identified at a taxonomic level other than ___domain. In order to account for the effects of sampling depth on alpha- and beta-diversity analyses, abundances were rarefied to the minimum sample read count greater than 10,000 reads. Therefore, OTU abundances were rarefied to 10,006 reads for saliva samples and 81,491 reads for stool samples. For beta-diversity analyses, KO abundances were rarefied to 10,141 reads for saliva samples, and 25,763 reads for stool samples.
Statistics and reproducibility
All statistical analyses were conducted in R version 4.1.244.
Covariate selection
To account for the influence of disease features on the microbiome diversity differences, we first examined which covariates were different between PD cases and healthy controls using non-parametric Wilcoxon rank-sum tests for numeric covariates and Chi-square tests for categorical covariates. We used a cutoff of α = 0.05 and did not adjust for multiple testing to include as many covariates as possible. We then tested whether the covariates that were different in PD cases and controls were correlated with two alpha-diversity metrics (Chao1 and Inverse Simpson) and two beta-diversity metrics (Sørensen and Bray–Curtis) for each microbiome source (oral and gut). We used Gaussian generalized linear models to assess the association of alpha-diversity with each covariate, and permutational ANOVA (PERMANOVA; 999 permutations) to determine differences in composition by each covariate. Covariates that differed in PD and were associated with alpha- or beta-diversity were candidates for inclusion in the remainder of the analyses. To avoid over-fitting our models, we chose only one covariate that best captured each clinical feature (e.g., number of weekly bowel movements, rather than Bristol stool scores 1, 4 and 6 for constipation). The final set of selected covariates was used for all statistical models, regardless of microbiome source. After QC and rarefaction steps, there was a small significant difference in age between cases and controls, therefore we included age as a covariate as well.
Subtyping symptom burden scores
To explore the association between the microbiome and disease severity, we built scoring functions based on self-reported answers from the Unified Parkinson’s Disease Rating Scale (part II; UPDRS)41. To assess the burden and severity of peripheral nerve involvement, we determined the presence/absence of two core features of autonomic dysfunction in PD. Subjects received a score of +2 if they reported constipation (defined as <3 bowel movements (BMs) weekly, difficulty moving bowels, or laxative use in the past month) and orthostatic hypotension, a score of +1 if they reported constipation or orthostatic hypotension, and a score of 0 if they reported neither. These features correspond to items 1.11 & 1.12 of the UPDRS part II.
Microbiome richness (alpha-diversity)
We chose Chao1 and inverse Simpson as the alpha-diversity metrics, as they provide different information regarding the diversity of the microbiome. Chao1 estimates the total richness of a community, agnostic to taxon abundances. Inverse Simpson, on the other hand, considers taxon abundance distributions, and provides an estimate of the size of an equivalently diverse community with equal abundance distribution. We used Wilcoxon rank sum tests to determine if alpha-diversity metrics differed between PD cases and controls, without inclusion of covariates, and used false discovery rate (FDR) correction of p-values for multiple tests. We used logistic regression to assess the ability of alpha-diversity metrics for oral and gut microbiomes to classify participants’ PD status. For each microbiome source (saliva and stool), we built two initial models, one for each alpha-diversity metric, that also included the covariates of interest. We assessed the models’ fit through a combination of visual inspection of binned residuals, Hosmer–Lemeshow goodness of fit tests, model R-squared values, and model Akaike information criteria (AICs). Using the model with the best fit, we tested the addition of interaction terms (alpha-diversity metric by a covariate of interest) using ANOVA to determine whether inclusion of the interaction term improved the model. We assessed significance at α = 0.05.
Differences in microbiome composition (beta-diversity)
As with alpha-diversity, we chose Sørensen and Bray–Curtis beta-diversity metrics because of their differential focus on changes in microbiome composition. Sørensen is a presence-absence metric that is sensitive to changes in rare taxa, while Bray–Curtis is abundance weighted, meaning it tends to detect shifts in the relative abundance of more common taxa. We used PERMANOVA models to assess the association between beta-diversity and PD status, and specifically the capscale function from the vegan R-package (version 2.6-4), which generates distance-based redundancy analysis (dbRDA) ordinations45. For each microbiome source (saliva and stool) and each microbiome feature type (OTUs and KOs), we built two initial models, one for each beta-diversity metric, that also included the covariates of interest. We assessed the models’ fit by comparing model R-squared values. Using the model with the best fit, we tested the addition of interaction terms (PD diagnosis by a covariate of interest) using ANOVA to determine whether inclusion of the interaction term improved the model. Additionally, we constructed dbRDA ordinations including counts from both saliva and stool samples (rarified to the saliva level indicated above of 10,006 reads per sample). We tested the significance of PD diagnosis in association with the beta-diversity metrics while conditioning on sample source to determine if there was an overall association between microbiome composition and PD diagnosis, regardless of sample source. The significance cutoff for all tests was α = 0.05, and all PERMANOVA tests utilized 999 permutations.
Differential abundance analyses
In order to identify microbial taxon and functional group indicators of PD case or controls, we conducted two separate differential abundance tests46. We constructed models using both the ancombc2 function (ANCOMBC R-package version 2.0.2)47 and the aldex.glm function (ALDEx2 R-package version 1.30.0)48. We identified as differentially abundant only those features with a FDR-adjusted p < 0.05 according to both tests. For each source (saliva or stool) and feature set (OTUs or KOs), we constructed two of these pairs of models, one pair which included only PD diagnosis as an independent variable (to compare directly to other similar analyses in the literature) and one pair that included PD diagnosis and selected covariates (to determine if the identification of certain features as indicators might be better explained by these covariates rather than PD diagnosis alone).
Network analysis
Microbial association networks (i.e., networks of abundance relationships among microbial features) were constructed using the function spiec.easi from the SpiecEasi R-package (version 1.1.2)49. Samples were split into cases or controls only and spiec.easi was run for both OTU and KO abundances. The spiec-easi method does not allow for covariate inclusion, and so this is the one analysis in which the selected covariates were not accounted for in the models. We assessed differences in network connectivity by comparing the average node degree (that is, the number of significant associations for each OTU or KO) between the control and case networks. Significant differences in average node degree were assessed using Wilcoxon rank-sum tests (α = 0.05 with FDR adjustment for multiple tests).
Random forest classification
We built random forest models for utilizing different combinations of taxonomic and functional abundances and survey responses to classify participants’ PD status. For all models, the participant population was split 70/30 into a training and test set, respectively, accounting for proportion of cases and controls in each split (65% cases, 35% controls). Feature (taxon and functional group) abundances were centered and log-ratio transformed. The ranger function (ranger R-package version 0.14.1)50 was used to build the random forest models. Hyperparameter tuning was conducted on each data set using 5-fold cross-validation. The tuned hyperparameters included “mtry” (number of variables to possibly split at in each node: six values chosen between 1 and the square root of the total number of features, rounded down), “trees” (number of trees in the forest: 10, 50, 100, 500, 1000, or 1500), and “splitrule” (the splitting rule: “gini” or “extratrees”). After tuning, models were fitted on the training data set and then used to predict the test data set. Model performance was assessed using the area under the curve (AUC) of the receiver operating characteristic (ROC) curve. These AUCs underwent Monte Carlo cross-validation by randomizing the training-testing split 99 times to confirm that the results from any single split were not due to the split itself.
The input data sets included, as a baseline, the survey answers selected as the covariates used in previous analyses as well as answers to additional survey questions that were significantly different between PD cases and controls. These additional survey questions included reported weekly frequency of Bristol stool scores 1, 4, and 6; breakfast consumption frequency; coffee consumption frequency; typical hours of sleep per night; and the report of three oral conditions: cavities, gingivitis, and receding gums. Additional input data sets included feature abundances at the OTU, species, genus, KO, module, and pathway levels. For each of these data sets, we constructed data sets that also included the selected survey answers. Furthermore, we compared models that included combinations of feature levels (e.g., OTU and species; or OTU, species, and genus abundances) with and without the covariates. Lastly, we assessed whether combining saliva and stool feature abundances improved the classification models as well. Relative model performance was determined by comparing mean ROC AUCs. An accounting of all the data sets compared can be found in Supplemental Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Participant characteristics
Supplementary Fig. 2 depicts the cohort sample sizes and the impact of QC and rarefaction on sample loss. The final sample size used for the primary analyses included 666 stool microbiome samples (445 PD cases and 221 controls) and 658 saliva microbiome samples (438 PD cases and 220 controls). The most represented states were California (10.6%), Texas (5.31%), Virginia (5.10%), and Colorado (4.00%). There was no influence of longitude or latitude, however geographic region (US census regions) did have a significant association with beta-diversity, and so was included as a covariate in all models. Inclusion of sequencing batch ID in analyses did not affect the interpretation of the results.
The mean age of the PD cohort was 61.7 years (interquartile range [IQR]: 58–66) with a slight male preponderance (54.6%). Mean age at diagnosis was 58.6 years (IQR: 55–63), and mean disease duration (time elapsed in years since self reported diagnosis) was 3.2 years (IQR: 2–4). The most frequently reported motor symptoms were tremor (65.8%), rigidity (40.4%), and bradykinesia (49.2%); 88.3% were treated with anti-parkinsonian medication, including dopamine precursors (70.6%), dopamine agonists (31.7%), and monoamine oxidase-B inhibitors (26.1%). We did not see any significant effect of medication class or number of PD medications currently prescribed on metrics of alpha-diversity. We observed a small effect of dopamine agonist use on beta-diversity for only the functional (KOs) profile of the saliva microbiome (Bray–Curtis: F = 2.37, FDR adj. p = 0.016; Sørensen: F = 2.29, FDR adj. p = 0.028). One-third (32.1%) of patients reported constipation. Controls were well-matched for both age and sex distribution (mean age 62.6 years; IQR 59–67.0; 56.8% male; Table 1).
Covariate features of PD
The covariates selected for inclusion in statistical models were age, BMI, dessert consumption frequency, ever having smoked, flossing teeth frequency, laxative use in the past month, number of weekly bowel movements and US census region. These terms were included in all alpha- and beta-diversity analyses and all differential abundance analyses. For the random forest classification, we included these covariates plus additional ones detailed previously in the methods.
Observed associations between PD and microbiome diversity and composition are robust to inclusion of covariates in models
Stool microbiome
We observed increased stool alpha-diversity in PD cases relative to healthy controls according to the Chao1 metric (W = 41,384; adj. p = 0.00173; Fig. 1a). The significant positive relationship between PD diagnosis and Chao1 remained after accounting for clinical covariates in the model (Odds per Chao1 S.D.: 1.0776, p = 0.008; Fig. 1b, see Supplementary Table 1 for diagnosis vs. covariates associations). Furthermore, we identified a significant interaction between PD diagnosis and number of weekly BMs, a metric of constipation, (LR Chisq = 8.51, p = 0.014; Supplementary Fig. 3a). Post-hoc Tukey tests confirmed that PD cases reporting fewer than 3 BMs per week had significantly higher alpha-diversity than controls (adj. p = 0.00339) and cases (adj. p = 0.0137) reporting regular (6 + ) BMs per week. The interaction between Chao1 and weekly BMs significantly improved the logistic regression model classifying participants into cases and controls (Dev. = 10.94, p = 0.00421). For participants reporting regular BM frequency, the odds ratio for Chao1 predicting PD cases probability was 1.08, but for participants reporting less than 3 BMs weekly, the Chao1 odds ratio was 2.55 (Supplementary Fig. 3B). This indicates that constipation symptoms in PD cases, but not controls, correlate with increased stool microbiome alpha-diversity.

Stool microbiome associations between PD diagnosis and alpha- and beta-diversity. For all plots, blue indicates controls and pink indicates PD cases. a Violin plot showing the Chao1 diversity metric for controls (n = 221) and cases (n = 445). The black error bars represent the 95% C.I.s around the means. b Regression plot showing statistically significant association between PD diagnosis and scaled Chao1 alpha-diversity metric estimated from a logistic regression model that also included the selected covariates for stool OTUs (n = 666). The black line indicates the estimated association and the shaded area represents the standard error around the estimate. Covariate p-values can be found in Supplementary Table 1. c dbRDA ordination showing statistically significant association between PD diagnosis and Sørensen beta-diversity metric estimated from a PERMANOVA model that also included the selected covariates for stool OTUs. The points indicate the centroids for controls (n = 221) and cases (n = 445) and the ellipses indicate 95% C.I.s of the centroids. CAP in the axes stands for “constrained analysis of proximities” and indicates the axis is constrained by input variables. Covariate p-values can be found in Supplementary Table 2. d dbRDA ordination showing statistically significant association between PD diagnosis and Bray–Curtis beta-diversity metric estimated from a PERMANOVA model that also included the selected covariates for stool KOs (controls n = 221, cases n = 445). The points, ellipses, and axes titles indicate the same as for c. Covariate p-values can be found in Supplementary Table 3.
For the stool microbiome taxonomic profile, we found a significant association between Sørensen distances and PD diagnosis (F = 3.73, p = 0.001; Fig. 1c; Supplementary Table 2), and for the functional profile, we found a significant association between Bray–Curtis distances and PD diagnosis (F = 1.43, p = 0.040; Fig. 1d; Supplementary Table 3). There was not a significant association between Bray–Curtis distances and PD diagnosis for the taxonomic profile, nor one between Sørensen distances and PD diagnosis for the functional profile. Interaction terms between PD diagnosis and the covariates did not improve the models’ performance to explain differences in beta-diversity. Thus there are significant differences in both taxonomic and functional composition of the stool microbiome between PD cases and controls, but these associations are not significantly altered by the disease-relevant covariates we assessed.
Symptom burden for the peripheral scores had a significant positive relationship with stool microbiome alpha-diversity (F = 5.64, adj. p = 0.037; Fig. 3a; Supplementary Table 4) as well as an association with significant differences in stool microbiome composition (F = 1.74, adj. p = 0.018; Fig. 3b; Supplementary Table 5). Taken together, these results suggest that the microbiome is most diverse in PD patients that have a peripheral involvement, and suggests a possible interplay between the severity of autonomic symptoms and microbial composition.
Saliva microbiome
We found significantly increased saliva microbiome alpha-diversity (inverse Simpson) in cases versus controls (W = 43,194; p = 0.030; Fig. 2a). However, when we constructed a logistic regression model to test the association between PD diagnosis and alpha-diversity, incorporating the disease-relevant covariates, we found no significant relationship (see Supplementary Table 6 for significant associations between PD diagnosis and covariates).

Saliva microbiome associations between PD diagnosis and alpha- and beta-diversity. For all plots, blue indicates controls and pink indicates PD cases. a Violin plot showing the inverse Simpson diversity metric for controls (n = 220) and cases (n = 438). The black error bars represent the 95% C.I.s around the means. b dbRDA ordination showing statistically significant association between PD diagnosis and Sørensen beta-diversity metric estimated from a PERMANOVA model that also included the selected covariates for saliva OTUs. The points indicate the centroids for controls (n = 220) and cases (n = 438) and the ellipses indicate 95% C.I.s of the centroids. Covariate p-values can be found in Supplementary Table 7. c dbRDA ordination showing statistically significant association between PD diagnosis and Sørensen beta-diversity metric estimated from a PERMANOVA model that also included the selected covariates for saliva KOs (controls n = 204, cases n = 399). The points and ellipses indicate the same as for (b). Covariate p-values can be found in Supplementary Table 8.
We identified significant differences in saliva microbiome composition (Sørensen) between controls and PD cases for both saliva OTUs (F = 3.35, p = 0.001; Supplementary Table 7) and saliva KOs (F = 1.79, p = 0.008; Supplementary Table 8) in models that included covariates. There were no significant associations between either the Bray–Curtis distances for either the taxonomic nor functional profiles of the saliva microbiome and PD diagnosis. We observed a significant interaction between PD diagnosis and teeth flossing frequency on metrics of saliva taxonomic beta-diversity (F = 1.53, p = 0.016; Supplementary Fig. 3c; Supplementary Table 5). In participants that “never flossed”, the taxonomic composition of the saliva microbiome was more distinct (i.e., a greater distance between group centroids in ordination space) between cases and controls compared to those that “flossed 1-2x/day”, suggesting that oral hygiene practices need to be accounted for when interpreting the microbiome of saliva in PD. For the functional composition of the saliva microbiome, we identified two covariates that significantly interacted with PD diagnosis: number of weekly BMs (F = 1.37, p = 0.028; Supplementary Fig. 3d), and US census region (F = 1.27, p = 0.041; Supplementary Fig. 3e). Similar to the flossing by diagnosis interaction, control and case participants reporting regular (6 + /week) BMs had more similar microbiome functional composition, while control and case participants reporting irregular (<3/week) BMs were much more distinct from each other. For participant geographic region, the interaction appears primarily driven by the fact that PD cases living in the west region have a functional composition more similar to controls from across the country than PD cases from the other three regions. These results highlight the importance of accounting for common PD disease features such as constipation, as well as demographics such as geographic ___location when interrogating the PD saliva microbiome.
There was no significant relationship between saliva microbiome alpha-diversity and peripheral symptom burden, but there were differences in composition between the UPDRS peripheral symptom burden levels (F = 1.95, adj. p = 0.018; Fig. 3c) for the PD saliva microbiome. This suggests that, while saliva microbiome diversity is not associated with peripheral symptom burden, there are significant changes in composition.

Association microbiome diversity metrics and severity of peripheral autonomic dysfunction for PD cases. a A violin plot of stool microbiome Chao1 scores for peripheral scores of no symptoms (green, n = 98) and orthostatic hypotension (OH) and constipation (orange, n = 22). b dbRDA ordination of Sørensen distances for stool microbiomes of PD participants with no peripheral burden (green, n = 92) or OH + constipation (orange, n = 22). Points show the group centroids and ellipses denote 95% C.I.s around the centroids. c dbRDA ordination of Sørensen distances for saliva microbiomes of PD participants with no peripheral burden (green, n = 95) or OH + constipation (orange, n = 21). Points show the group centroids and ellipses denote 95% C.I.s around the centroids.
Disrupted microbiome association networks in PD
To investigate the degree of microbiome disruption in PD, we explored microbe-microbe and function-function co-abundance networks in cases and controls. In both cohorts, the saliva OTUs had, on average, more significant associations compared to the stool OTUs (Fig. 4), which could be due to the greater number of OTUs detected in the saliva versus the stool microbiome. Using microbe-microbe network analyses, compared to healthy controls, PD cases had more disrupted networks (i.e., lower average number of connections per OTU) and fewer total significant associations in stool, but not saliva (Fig. 4). In stool, the average degree for individual microbes was significantly lower in the PD cases (W = 138,340; FDR-adjusted p = 2.73 × 10−7). For the function-function networks, we found PD cases were disrupted compared to healthy controls for both the saliva microbiome (W = 1,297,487; FDR-adjusted p = 0.019), and the stool microbiome (W = 1,348,038; FDR-adjusted p = 0.00186; Supplementary Fig. 4). Additionally, we assessed the microbe-microbe co-abundances between the saliva and stool microbiomes. Compared to controls, the between-source network had many more un-associated (no significant co-abundances) OTUs in PD cases (Fig. 5). Together, these findings suggest that while the diversity of the microbiome is increased in PD cases, their microbial networks have lost connections.

Microbe-microbe (OTU level) abundance association networks for saliva and stool, and controls and PD cases. The networks in the top row are for stool OTUs in controls (a) and PD cases (b). The networks on the bottom row are for saliva OTUs in controls (d) and PD cases (e). Each point represents an individual OTU, and the size of the point indicates its degree centrality metric. The colored points are for OTUs that have a top 10 degree metric for that specific community (e.g., saliva controls), to highlight the most connected taxa in each network. The color within these points represents the OTU’s taxonomic family. The lines connecting points indicate statistically significant associations in their abundances as determined by the spiec-easi method. The lines are colored and scaled by the absolute value of their correlation coefficient with darker and wider indicating greater correlation magnitude. Solid lines indicate positive correlations, dashed lines indicate negative correlations. The density plots to the right of the networks, for stool (c) and saliva (f) show the distributions of degree centralities for each OTU (node) in the networks. The lines indicate the mean degree for controls (blue) and cases (pink). An asterisk indicates a significant difference in degree between controls and cases, and an “n.s.” indicates no significant difference.

Microbe-microbe (OTU level) abundance association networks between saliva and stool for controls and PD cases. a The network for controls. b The network for PD cases. Each point represents an individual OTU, and the size of the point indicates its degree centrality metric. The colored points are for OTUs that have a top 10 degree metric for that specific community (e.g., saliva controls), to highlight the most connected taxa in each network. The color within the point represents the OTU’s taxonomic family and its shape indicates whether it was sampled from the saliva (circle) or stool (square). The lines connecting points indicate statistically significant associations in their abundances as determined by the spiec-easi method. The lines are colored and scaled by the absolute value of their correlation coefficient with darker and wider indicating greater correlation magnitude. Solid lines indicate positive correlations, dashed lines indicate negative correlations. c A density plot showing the distributions of degree centralities for each OTU (node) in the networks.
Differentially abundant microbiome features in PD cases versus controls
In order to understand the granular differences in the microbiome between PD cases and controls, we identified OTUs and KOs that were differentially abundant between the two cohorts.
Stool microbiome differential abundances
We identified a number of differentially abundant stool microbiome OTUs and KOs (Fig. 6). With the addition of the covariates, we found two OTUs that were indicators of healthy controls (Parabacteroides distasonis, and Ruminococcus gnavus) and five OTUs that were indicators of PD cases (Burkholderiales order, Ruminococcaceae family, Bifidobacterium dentium, Streptococcus vestibularis OTU, and one Anaerostipes hadrus OTU; Fig. 6a). Of note, taxa commonly reported as elevated in PD, such as Akkermansia, or depleted in PD, such as Roseburia, were significantly differentially abundant only when we did not include any covariates in the models (Supplementary Fig. 5a).

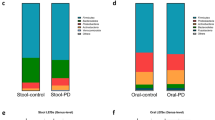
Significant differentially abundant OTU-level microbial taxa. Bars indicate centered-log transformed log2-fold difference in abundance between controls (toward left [negative], n = 221) and PD cases (towards right [positive], n = 445). Significance was assessed with two tests (ANCOMBC2 and ALDEx2). a Taxa from the stool microbiome. b Taxa from the saliva microbiome. Taxa are ordered by relative differential abundance and the bars are colored by taxonomic family (controls n = 220, cases n = 438). Exact p-values for all OTUs can be found in Supplemental Data 2.
At a functional level, we identified a single significant KO indicator of PD status that remained significant after accounting for disease-related covariates (Supplementary Fig. 5b), which was involved in membrane transport (beta−glucoside PTS system EIICBA component).
Saliva microbiome differential abundances
Differential abundance testing of the saliva microbiome identified fifteen Streptococcus OTUs that were indicative of either cases or controls. For example, three S. mutans OTUs, one S. vestibularis OTU, and one unspecified Streptococcus species were elevated in PD cases, while ten others, primarily unspecified Streptococcus species plus one S. australis and two S. pseudopneumoniae OTUs, were depleted in PD relative to controls (Fig. 6b). Four Actinomyces OTUs had higher relative abundance in PD cases, while two were higher in controls, and none of these six OTUs were identified to the same Actinomyces species. All four Neisseria OTUs and the one Prevotella loescheii OTU that were significantly differentially abundant were depleted in PD cases relative to controls.
With regard to the functional profile of the saliva microbiome, we identified six significant KO indicators of cases, which were not significant after including the covariates. Of these six, two were involved in carbohydrate metabolism (Supplementary Fig. 6b). Two other KOs were involved with response to antibiotics: penicillin−binding protein 2 A, which binds to beta-lactam antibiotics, and a sensor histidine kinases (LiaS), which has been functionally linked to cell wall-active antibiotics that interfere with the lipid II cycle in the cytoplasmic membrane51. One KO, pertactin, is a virulence factor of Bordetella pertussis, though no OTUs in this cohort were assigned to any Bordetella species.
The full results of all taxa and functional groups tested for differential abundance can be found in Supplemental Data 2, and the numbers of features (OTUs and KOs) that reached FDR < 0.05 per analysis (ANCOMBC2, ALDEx2, or both) for all data sets can be found in Supplementary Table 9.
Saliva microbiome OTU profiles improve machine learning classification of PD cases versus controls
To identify microbial differences that discriminated between cases and controls, we constructed a series of random forest classifier models to predict participants’ PD status. First, we determined how well covariates (survey answers) that were significantly different between cases and controls performed alone. In addition to the survey answers used in the previous analyses, we included responses regarding cavities, gingivitis, receding gums, coffee consumption habits, and hours of sleep per night. The survey answers alone did better than chance at classifying participants’ PD diagnosis (mean ROC AUC = 0.726; Fig. 7).

Model performance for random forest classifiers. Random forest models were built using a variety of input data sets. As a baseline, answers to survey questions were used. The points above represent the mean ROC AUCs and the line segments represent the standard error around these means. The dashed vertical line represents the mean ROC AUC for the baseline data set, i.e., the survey answers alone. The white points indicate the mean ROC AUCs for the saliva taxonomic abundances, and the black points for the stool taxonomic abundances. The y-axis identifies the data sets included in these analyses, and the bracket highlights the two best performing models with and without covariates in the data set.
For single-level taxonomic profiles, the OTU- and species-level abundances of the saliva microbiome generated the best performing models (saliva OTU mean AUC = 0.758, saliva species mean AUC = 0.745; Fig. 7; Supplementary Table 1). The addition of survey answers improved performance, and the top three overall models all included both saliva OTU abundances and the survey answers (surveys + OTUs + species mean AUC = 0.776; surveys + OTUs + species + genera mean AUC = 0.776; surveys + OTUs mean AUC = 0.770). The functional profiles for both the stool and saliva microbiomes did not offer any advantages over the survey answers alone (Supplementary Table 1). For the best performing model, the ten most important features were microbial taxa, with Prevotella loescheii having the highest importance score. The survey answer with the highest importance score was the number of weekly BMs reported by the participants (Supplementary Fig. 7 and Supplementary Data 3).
Discussion
To our knowledge, this is the largest saliva and stool microbiome study of PD, utilizing shotgun metagenomic sequencing from a geographically dispersed US-based population with well-matched controls. As shown recently, matching of control subjects is necessary in human microbiome research to appropriately account for potentially confounding host factors52. Our results support earlier studies that stool and saliva microbiomes of PD patients differ from healthy control subjects, exhibiting overall higher alpha-diversity. Several differentially abundant microbiome taxa and shotgun metagenomic-resolved functions were robust statistical predictors of PD, and were not explained by other common disease features. A novel finding of our study is the potential value of saliva OTU abundances, which provided the best discrimination between PD cases and controls.
Our first aim was to replicate findings of previous studies and determine which were robust indicators of diagnosis and not better explained by disease-associated features that are more common in PD, but also occur in healthy participants due to other factors. Increased alpha-diversity of PD cases versus controls was more robust in stool versus saliva, and remained significant even after controlling for disease-relevant covariates. This result is in line with previous literature, in which many (but not all) studies report greater stool microbiome richness in PD cases versus controls5,30,32,34, whereas, for the saliva microbiome, alpha-diversity is often reported as not significantly different32,36,37, or studies conflict as to whether it is greater or lower15,35. Increased gut alpha-diversity in PD appears to be a finding not solely explainable by covariates such as constipation. Cases reporting constipation (<3 BMs/week) had higher alpha-diversity than those reporting 6+ BMs/week, but this association was not observed among controls (Supplementary Fig. 3). This suggests that constipation has a different impact on the microbiome in PD cases than in controls. To explore this idea further, we examined the association between PD microbiome diversity and peripheral autonomic nervous system function by comparing alpha-diversity in patients with and without autonomic symptoms. Greater peripheral symptom severity (OH and constipation) positively correlated with stool alpha-diversity. This suggests that stool microbiome perturbation correlates with the burden of peripheral efferent nerve involvement, indicating a concerted peripheral PD phenotype with connections between the enteric nervous system and the microbiome.
In both the stool and saliva, beta-diversity measured by Sørensen distance best distinguished PD cases from controls. Because this metric only considers taxon presence or absence, this result implies that the presence or absence of rare taxa as opposed to shifts in the relative abundances of common taxa are important microbiome differences between PD patients and healthy individuals. Previous studies have fairly consistently reported significant differences in composition between controls and PD cases for both stool and saliva microbiomes5,15,30,32,34,35,36,46. For saliva, there was also a significant interaction between PD diagnosis and the frequency with which participants reported flossing. We tested this interaction specifically because, due to the burden of bradykinesia and resting tremor in PD participants, fine motor tasks such as those required for oral hygiene can be more difficult than for people without those symptoms. We expected healthy controls that did not floss regularly to have saliva microbiome compositions more similar to PD cases that also did not floss regularly. However, contrary to our expectations, among participants who reported never flossing, the differences in composition between PD cases and controls were greater than among participants who reported flossing regularly. This implies that some other feature of PD is likely driving differences in the composition of the saliva microbiome between cases and controls.
We wanted to determine whether the co-abundance networks of metagenomic-resolved taxa and metabolic function (KOs) in healthy control microbiomes were maintained in PD microbiomes, despite differences in composition. We assessed the degree centrality metric for each OTU and KO node in the networks and found decreased connectivity of taxa in the stool microbiome and decreased connectivity of KOs in both stool and saliva. So, while alpha-diversity was increased in the PD microbiomes compared to controls, the co-abundance networks were disrupted. Additionally, we found that the significant co-abundance relationships in PD cases were not a subset of those in the controls (only 29.7% of the total significant co-abundances were shared by both cohorts). This implies that the gut ecology of PD is distinct from that of controls, that there are fewer synergistic interactions in these communities in PD, and that more taxa are operating independently of others.
In addition to summary diversity metrics, we wanted to understand whether there were significant differences in individual taxon and functional group abundances between controls and cases. For the stool microbiome, we noted with interest that Akkermansia (genus) was significantly elevated in PD cases only when covariates were not included in the model. Akkermansia is one of the most consistently elevated genera in PD literature10,11,21; however, it appears that its differential abundance may be better explained by at least one of the covariates we considered. Indeed, this particular Akkermansia OTU negatively correlated with BMI, regardless of PD diagnosis (Supplementary Table 10). Similarly, the genus Roseburia is commonly reported as depleted in PD relative to controls10,11,21, but we found that the abundance of one Roseburia OTU was significantly decreased in participants reporting fewer than 6 bowel movements weekly and those reporting laxative use in the past month (Supplementary Table 11).
Other OTUs within genera such as Bifidobacterium and Streptococcus, which have been reported as commonly elevated in PD, and OTUs within genera such as Ruminococcus and Parabacteroides, which have been reported as depleted in PD10,11,21,33, were significantly differentially abundant between cases and controls even when covariates were included in the models. These taxa appear to be more directly associated with PD-specific factors rather than less specific symptoms such as constipation.
Previous research has identified various microbial cell transporters as indicative of PD32, and one of the two KOs significantly elevated in PD versus controls in the model that included covariates specifically involved functional groups associated with transport of sugars across cellular membranes (beta−glucoside PTS system EIICBA component).
For the saliva microbiome, there was much less within-genus congruence regarding differential abundances between cases and controls. This may explain, in part, why there has been less consensus in the literature regarding saliva microbiome PD indicators, as much work to date has utilized 16S amplicon sequencing, which is often only reliably annotated to the genus level. For example, within the Streptococcus genus, we identified a number of OTUs indicative of either cohort, which is congruent with the fact that it is a genus that includes taxa with widely varied functions53, unlike some genera which are much more homogenous. Of particular note were three OTUs of the species S. mutans that were particularly elevated in PD cases. S. mutans is a primary contributor to tooth decay54, and its elevated abundance in cases might indicate that it is simply a marker of reduced oral hygiene from difficulty with brushing or flossing due to the motor symptoms associated with PD. However, multiple OTUs within the genera Neisseria and Fusobacterium were found to be depleted in PD. Multiple Neisseria taxa have also been implicated in contributing to tooth decay, and the species F. periodonticum can form oral biofilms along with relatives such as F. nucleatum, which is a known opportunistic pathogen that can contribute to periodontal disease. The OTU that was most depleted in PD cases belonged to the species Prevotella loescheii has been recovered from oral infections like periodontitis55. Thus, it is not obvious that the differential abundances in oral OTUs observed between cases and controls can be explained by differences in oral hygiene. Previous work has shown that PD cases tend to have elevated levels of IL-6 in their gingival crevicular fluid15, and Neisseria has been shown to have a negative association with oral levels of IL-656. Features of the disease such as elevated levels of pro-inflammatory cytokines15 or excess saliva in the mouth due to lack of swallowing36 may be contributing to this pattern of oral OTU abundances in PD cases versus controls, and warrant further investigation.
Even fewer published studies have assayed the functional profile of the saliva microbiome. One such study identified functional groups related to the citrate cycle and tryptophan metabolism as depleted in PD cases, and functional groups related to metabolism (glycolysis and the pentose phosphate pathway) and microbial formaldehyde assimilation as elevated37. In our study, none of the KOs assessed reached the significance threshold in models that included covariates. However, for the models that only considered diagnosis, there were six differentially abundant KOs, two of which were involved in carbohydrate metabolism, specifically of simple sugars such as glucose and sucrose. Additionally, we identified KOs involved with response to antibiotics as elevated in PD saliva. Previous work has indicated that antibiotic resistance genes have greater abundance in PD patients’ stool32, and certain types of oral antibiotics have been shown to be correlated with an elevated risk of PD57. Our exclusion criteria included no antibiotic use in the past 6 months, but even earlier antibiotic use could potentially contribute to these results.
Finally, we wanted to determine how informative microbial abundances were for classifying samples by diagnosis. The best performing random forest models all included saliva microbiome OTU abundances. Stool genus abundances did outperform saliva genus abundances, which has been shown previously32; however, these models were nowhere near the top performance of all the models we assessed. The range of AUCs for the model utilizing stool taxon abundances (0.68–0.75) compares similarly to other studies examining the microbiome in early PD (0.69–0.76)5. Previous work has also shown that OTU-level abundances from transcriptomic sequencing of the saliva microbiome perform well (AUC > 0.9; logistic regression) at classifying PD cases versus controls, but this performance was not compared to that of stool OTU-level abundances, and was based on only 84 total participants37. Firstly, this result highlights the importance of utilizing sequencing methods that can reliably identify microbial taxa to sub-species levels when studying the PD microbiome. Secondly, this suggests that future research into identifying early PD might benefit from including the saliva and stool microbiotic profiles in conjunction with other biomarkers findings. Because saliva is much less onerous (for participants and researchers alike) to sample than is stool or blood, saliva-based analyses of PD biomarkers could include many more participants and allow for more repeat sampling in longitudinal studies.
As with all studies, there are particular limitations to our findings. Firstly, PD diagnosis was self-reported, but we have shown excellent concordance between self-reporting and a validation study of diagnosis by trained neurologists58. Similarly, clinical features were assessed by subjective survey answers rather than objective clinical assessments. There may also be inaccuracies in determining if the participants’ orthostatic symptoms were related to hypotension, as we did not have blood pressure readings. While we purposely recruited participants across the entirety of the United States, this study was still restricted to European ancestry living within the US, and may not be representative of more diverse populations within the US and other countries. Further studies are needed to explore the impact of dopaminergic medication class or dosage on the microbiota. Despite our best efforts, there was a small (0.9 years), but significant difference in age between the controls and cases. While we do not believe this accounts for the magnitude of the microbiome differences observed, we used age as a covariate in our statistical models. Lastly, as this is a cross-sectional observational study, it cannot provide information on causality, and the cohort size is underpowered for causal inference methods such as Mendelian randomization.
With this study, we confirmed significant differences in composition as well as connectivity of both stool and saliva microbiomes between PD cases and controls. We have identified taxa that robustly discriminate between these cohorts and are strong candidates for further research into their potential roles in the development of PD or their contributions to PD symptoms. Lastly, we suggest that interrogation of the PD saliva microbiome might yield valuable insights into disease development, diagnosis, and progression.
Data availability
Sequencing data, OTU and KO annotation tables, and all participant data underlying the figures and tables in this article are available through the Fox DEN tool (https://foxden.michaeljfox.org). To access these data, researchers are asked to create an account and to complete and e-sign a data use agreement (DUA) at https://foxden.michaeljfox.org/insight/register/. The DUA stipulates that access to the data will be granted for researchers via a non-exclusive, non-sublicensable, non-transferable license to access, store, use and disclose the data. A brief explanation of what the researcher intends to use the data for must be provided as well, which must involve research into Parkinson’s disease and closely related disorders, and not for any other purpose such as the diagnosis or treatment of human subjects.
Code availability
The code used to run the analyses can be found at https://github.com/23andMe/PD-microbiome59.
References
Saper, C. B. The central autonomic nervous system: conscious visceral perception and autonomic pattern generation. Annu. Rev. Neurosci. 25, 433–469 (2002).
Browning, K. N. Stress‐induced modulation of vagal afferents. Neurogastroenterol. Motil. 31, e13758 (2019).
Powley, T. L. et al. Vagal innervation of the stomach reassessed: brain−gut connectome uses smart terminals. Ann. N. Y. Acad. Sci. 1454, 14–30 (2019).
Heintz-Buschart, A. et al. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 33, 88–98 (2018).
Palacios, N. et al. Metagenomics of the gut microbiome in Parkinson’s disease: prodromal changes. Ann. Neurol. 94, 486–501 (2023).
Huang, B. et al. Gut microbiome dysbiosis across early Parkinson’s disease, REM sleep behavior disorder and their first-degree relatives. Nat. Commun. 14, 2501 (2023).
Yoon, H. J. et al. Dietary quality and the gut microbiome in early-stage Parkinson’s disease patients. Nutr. Neurosci. 1–9 https://doi.org/10.1080/1028415X.2023.2253025 (2023).
Del Tredici, K. & Braak, H. Review: Sporadic Parkinson’s disease: development and distribution of α-synuclein pathology. Neuropathol. Appl. Neurobiol. 42, 33–50 (2016).
Nie, S. & Ge, Y. The link between the gut microbiome, inflammation, and Parkinson’s disease. Appl. Microbiol. Biotechnol. 107, 6737–6749 (2023).
Berthouzoz, E. et al. Oral and intestinal dysbiosis in Parkinson’s disease. Rev. Neurol. (Paris) https://doi.org/10.1016/j.neurol.2022.12.010 (2023).
Klann, E. M. et al. The gut–brain axis and its relation to Parkinson’s disease: a review. Front. Aging Neurosci. 13, 782082 (2022).
Challis, C. et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 23, 327–336 (2020).
Stokholm, M. G., Danielsen, E. H., Hamilton‐Dutoit, S. J. & Borghammer, P. Pathological α‐synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 79, 940–949 (2016).
Lebouvier, T. et al. Pathological lesions in colonic biopsies during Parkinson’s disease. Gut 57, 1741–1743 (2008).
Fleury, V. et al. Oral dysbiosis and inflammation in Parkinson’s disease. J. Park. Dis. 11, 619–631 (2021).
Walsh, J., Griffin, B. T., Clarke, G. & Hyland, N. P. Drug-gut microbiota interactions: implications for neuropharmacology. Br. J. Pharmacol. 175, 4415–4429 (2018).
Hirayama, M., Nishiwaki, H., Hamaguchi, T. & Ohno, K. Gastrointestinal disorders in Parkinson’s disease and other Lewy body diseases. Npj Park. Dis. 9, 71 (2023).
Mateo, D., Marquès, M., Domingo, J. L. & Torrente, M. Influence of gut microbiota on the development of most prevalent neurodegenerative dementias and the potential effect of probiotics in elderly: a scoping review. Am. J. Med. Genet. B Neuropsychiatr. Genet. e32959 https://doi.org/10.1002/ajmg.b.32959 (2023).
Kalecký, K. & Bottiglieri, T. Targeted metabolomic analysis in Parkinson’s disease brain frontal cortex and putamen with relation to cognitive impairment. Npj Park. Dis. 9, 84 (2023).
Boertien, J. M., Pereira, P. A. B., Aho, V. T. E. & Scheperjans, F. Increasing comparability and utility of gut microbiome studies in Parkinson’s disease: a systematic review. J. Park. Dis. 9, S297–S312 (2019).
Li, Z. et al. Gut bacterial profiles in Parkinson’s disease: a systematic review. CNS Neurosci. Ther. cns.13990 https://doi.org/10.1111/cns.13990 (2022).
Lin, C.-H. et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflammation 16, 129 (2019).
Hasegawa, S. et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLOS One 10, e0142164 (2015).
Tan, A. H. et al. Gut microbial ecosystem in Parkinson disease: new clinicobiological insights from multi-omics. Ann. Neurol. 89, 546–559 (2021).
Barichella, M. et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 34, 396–405 (2019).
Hill-Burns, E. M. et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome: PD, Medications, and Gut Microbiome. Mov. Disord. 32, 739–749 (2017).
Scheperjans, F. et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015).
Cirstea, M. S. et al. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov. Disord. 35, 1208–1217 (2020).
Petrov, V. A. et al. Analysis of gut microbiota in patients with Parkinson’s disease. Bull. Exp. Biol. Med. 162, 734–737 (2017).
Baldini, F. et al. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 18, 62 (2020).
Aho, V. T. E. et al. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol. Neurodegener. 16, 6 (2021).
Jo, S. et al. Oral and gut dysbiosis leads to functional alterations in Parkinson’s disease. Npj Park. Dis. 8, 1–12 (2022).
Wallen, Z. D. et al. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 13, 6958 (2022).
Toh, T. S. et al. Gut microbiome in Parkinson’s disease: new insights from meta-analysis. Parkinsonism Relat. Disord. 94, 1–9 (2022).
Zapała, B. et al. The role of the western diet and oral microbiota in Parkinson’s disease. Nutrients 14, 355 (2022).
Rozas, N. S., Tribble, G. D. & Jeter, C. B. Oral factors that impact the oral microbiota in Parkinson’s disease. Microorganisms 9, 1616 (2021).
Mihaila, D. et al. The oral microbiome of early stage Parkinson’s disease and its relationship with functional measures of motor and non-motor function. PLOS One 14, e0218252 (2019).
Pereira, P. A. B. et al. Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat. Disord. 38, 61–67 (2017).
Arıkan, M. et al. Metaproteogenomic analysis of saliva amples from Parkinson’s Disease patients with cognitive impairment. npj Biofilms Microbiomes 9, 1–10 (2023).
Harvey, S., Matthai, S. & King, D. A. How to use the Bristol Stool Chart in childhood constipation. Arch. Dis. Child. Educ. Pract. Ed. 108, 335–339 (2023).
Goetz, C. G. et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov. Disord. 23, 2129–2170 (2008).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinforma. Oxf. Engl. 30, 2068–2069 (2014).
R Core Team. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, Vienna, Austria, 2021).
Oksanen, J. et al. Vegan: Community Ecology Package. https://CRAN.R-project.org/package=vegan (2022).
Wallen, Z. D. Comparison study of differential abundance testing methods using two large Parkinson disease gut microbiome datasets derived from 16S amplicon sequencing. BMC Bioinform. 22, 265 (2021).
Lin, H. & Peddada, S. D. Multigroup analysis of compositions of microbiomes with covariate adjustments and repeated measures. Nat. Methods 21, 83–91 (2024).
Fernandes, A. D. et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15 (2014).
Kurtz, Z. D. et al. Sparse and compositionally robust inference of microbial ecological networks. PLOS Comput. Biol. 11, e1004226 (2015).
Wright, M. N. & Ziegler, A. ranger: a fast implementation of random forests for high dimensional data in C++ and R. J. Stat. Softw. 77, 1–17 (2017).
liaS - Sensor histidine kinase LiaS - Bacillus subtilis (strain 168) | UniProtKB | UniProt. https://www.uniprot.org/uniprotkb/O32198/entry.
Vujkovic-Cvijin, I. et al. Host variables confound gut microbiota studies of human disease. Nature 587, 448–454 (2020).
Gao, X.-Y., Zhi, X.-Y., Li, H.-W., Klenk, H.-P. & Li, W.-J. Comparative genomics of the bacterial genus streptococcus illuminates evolutionary implications of species groups. PLoS One 9, e101229 (2014).
Loesche, W. J. Microbiology of dental decay and periodontal disease. in Medical Microbiology (ed. Baron, S.) (University of Texas Medical Branch at Galveston, 1996).
Könönen, E., Fteita, D., Gursoy, U. K. & Gursoy, M. Prevotella species as oral residents and infectious agents with potential impact on systemic conditions. J. Oral Microbiol. 14, 2079814 (2022).
Sarkar, A., Kuehl, M. N., Alman, A. C. & Burkhardt, B. R. Linking the oral microbiome and salivary cytokine abundance to circadian oscillations. Sci. Rep. 11, 2658 (2021).
Mertsalmi, T. H., Pekkonen, E. & Scheperjans, F. Antibiotic exposure and risk of Parkinson’s disease in Finland: a nationwide case‐control study. Mov. Disord. 35, 431–442 (2020).
Dorsey, E. R. et al. Virtual research visits and direct-to-consumer genetic testing in Parkinson’s disease. Digit. Health 1, 2055207615592998 (2015).
Stagaman, Keaton “23andMe/PD-Microbiome: first release”. Zenodo https://doi.org/10.5281/ZENODO.13647334 (2024).
Acknowledgements
We would like to thank the research participants and employees of 23andMe for making this work possible. We thank The Michael J. Fox Foundation for Parkinson’s Research for funding this research (K.S., M.J.K., and L.N.K.).
Author information
Authors and Affiliations
Consortia
Contributions
K.S. conducted all statistical analyses and was the primary author on the manuscript. M.J.K., S.A., T.F.S., P.F., and M.V.H. provided feedback on the analyses and aided in the editing of the manuscript. S.T.W., M.C.H., and L.N.K. helped K.S. in developing the analysis plan, provided input on the structure of the manuscript, and aided in the editing of the manuscript. J.T. approved the analysis plan and drafting of the manuscript. 23.R.T. provided access to the data and the infrastructure to make the analyses possible.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: K.S., M.J.K., M.W., S.A., T.F.S., P.F., J.T., M.V.H., and L.N.K. are employed by and hold stock or stock options in 23andMe, Inc.
Peer review
Peer review information
Communications Medicine thanks Haydeh Payami and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Stagaman, K., Kmiecik, M.J., Wetzel, M. et al. Oral and gut microbiome profiles in people with early idiopathic Parkinson’s disease. Commun Med 4, 209 (2024). https://doi.org/10.1038/s43856-024-00630-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43856-024-00630-8
This article is cited by
-
Metagenomic analysis reveals microbial drivers of heat resistance in dairy cattle
Animal Microbiome (2025)
-
Altered oral health and microbiota in drug-free patients with schizophrenia
BMC Psychiatry (2025)
