
Abstract
Interferon Regulatory Factors (IRFs), a family of transcription factors, profoundly influence the immune system, impacting both physiological and pathological processes. This review explores the diverse functions of nine mammalian IRF members, each featuring conserved domains essential for interactions with other transcription factors and cofactors. These interactions allow IRFs to modulate a broad spectrum of physiological processes, encompassing host defense, immune response, and cell development. Conversely, their pivotal role in immune regulation implicates them in the pathophysiology of various diseases, such as infectious diseases, autoimmune disorders, metabolic diseases, and cancers. In this context, IRFs display a dichotomous nature, functioning as both tumor suppressors and promoters, contingent upon the specific disease milieu. Post-translational modifications of IRFs, including phosphorylation and ubiquitination, play a crucial role in modulating their function, stability, and activation. As prospective biomarkers and therapeutic targets, IRFs present promising opportunities for disease intervention. Further research is needed to elucidate the precise mechanisms governing IRF regulation, potentially pioneering innovative therapeutic strategies, particularly in cancer treatment, where the equilibrium of IRF activities is of paramount importance.
Similar content being viewed by others


Introduction
Interferon regulatory factors (IRFs) constitute a comprehensive category of transcription factors initially identified as regulators of type I interferon (IFN-I) and IFN-responsive genes, and their mechanisms have been extensively researched for over the past 30 years. To date, notable progresses have been made in elucidating the multifaceted and pivotal role of IRFs within the homeostatic defense mechanisms of the host and in orchestrating immune responses to both internal and external stimuli; notably, they are instrumental in enhancing antiviral responses, provoking pro-inflammatory reactions, and cell development and differentiation.1,2,3 Furthermore, IRF family members have been recognized to harbor dual roles in immunity, exhibiting both anticancer and cancer-promoting properties.1 IRF family members are implicated across a spectrum of human pathologies, encompassing infectious diseases, autoimmune and inflammatory disorders, metabolic dysfunctions, and oncogenesis.4,5,6,7,8,9
In mammals, nine IRF members have been reported,10 designated as IRF1 (also known as MAR), IRF2, IRF3, IRF4 (known alternatively as LSIRF/ICSAT/ PIP), IRF5, IRF6, IRF7, IRF8 (also termed as ICSBP), and IRF9 (also named as ISGF3γ or P48), each characterized by conserved multi-___domain structures. Furthermore, an additional pair of IRF members, IRF10 and IRF11, have been discovered in avian species (IRF10) and teleost fish (IRF10 and IRF11), but these are notably absent in human and murine genomes.11,12 The inaugural member of the IRF family, IRF1, was unveiled by the Taniguchi laboratory in 1988, and it has been documented to facilitate virus-induced transcription by engaging with IFN-β enhancer elements.13,14 Later, Taniguchi’s group isolated the cDNA of IRF2 in 1989 by cross-hybridization with IRF1 cDNA.15 The IRF2 gene exhibits significant homology with the IRF1 gene within the 5’ portion of the protein-coding region.15 Additionally, IRF2 acts as an antagonist to IRF1, competing for the same promoter elements of IFN-I and IFN-II-inducible genes, thus potentially suppressing IRF1 function in specific contexts.15 In 1990, Driggers PH et al. characterized the IFN consensus sequence-binding protein (ICSBP), now identified as IRF8, initially cloned as a protein regulated by IFN-γ with a binding affinity for the IFN-inducible enhancer of major histocompatibility complex (MHC) class I genes.16 Moreover, Driggers PH and colleagues elucidated that ICSBP serves as a negative regulator, repressing the transcription of target genes activated by IFN or IRF1.17 Due to its derivation from a component of the IFNα-stimulated transcription factor 3 (ISGF3) complex, IRF9 was initially designated as ISGF3γ in 1990.18 ISGF3 constitutes a multiprotein complex comprised of four discrete polypeptides with molecular weights of 113, 91, 84, and 48 kDa, respectively.19 The 48 kD subunit is denoted as ISGF3γ, its expression level being upregulated by IFN-γ.19,20 ISGF3γ can associate with ISGF3α subunits, which are activated from a latent cytosolic form by IFN-I, to mediate antiviral activities. In 1995, Pitha group identified a novel IRF family member, IRF3, by mining homologs of IRF1 and IRF2 within an EST database, which associates with the IFN-stimulated response element (ISRE) to activate the expression of IFN-induced genes21 Furthermore, the Pitha group initially reported that IRF3 might potentiate transcription by forming complexes with other transcription factors, possibly members of the signal transducer and activator of transcription (STAT) family.21 In the same year, Matsuyama T. and colleagues identified a novel member of the IRF family, initially called the lymphoid-specific member of the IRF (LSIRF) and subsequently renamed IRF4.22,23 The cDNA for LSIRF was initially cloned from mouse spleen tissue, encoding a protein with 51 kDa molecular weight.22 IRF4 has been characterized using various terms in different contexts: as PU.1 interacting partner (PIP), it is a lymphoid-specific protein that binds to the enhancer elements of immunoglobulin light-chain genes, contingent on the presence of PU.1;24 as ICSAT, the human homolog of PIP and LSIRF, is isolated from adult T-cell leukemia cells or activated T cells.25 Distinctly, IRF4 differs from other IRF members as it is the sole IRF factor that is not regulated by IFNs.22 IRF4 induction can occur through a variety of antigen receptor-mediated stimuli, including plant lectins, CD3, and IgM cross-linking. Its function as a transcriptional activator or repressor is dictated by its interactions with an array of transcription factors or specific DNA-binding motifs22,26 IRF5 and IRF6, sharing structural homology, were first identified as members of the IRF family through the GeneBank database (accession numbers: human IRF5, U51127; human IRF6, AF027292).27 In 2001, Pitha group first demonstrated that IRF5 participated IFN-I gene induction.28 Prior research has established that IRF6 is predominantly engaged in developmental processes rather than in IFN gene expression, with its mutations being implicated in genetic disorders such as van der Woude syndrome, characterized by orofacial clefts and skin abnormalities.29,30 Likewise, IRF7 was initially identified as a novel constituent of the IRF family through the GeneBank database (accession numbers U73036, U73037) and information on IRF7 can be traced back to 199710,31 The IRF7 cDNA was cloned by a yeast one-hybrid system, encoding proteins that interact with sequences in the Epstein-Barr virus (EBV) BamHI Q promoter.31 Exhibiting the greatest amino acid homology with IRF3, IRF7 possesses the ability to bind to the ISRE sequence, thereby inhibiting transcriptional activation mediated by both IFN and IRF1.10,31
In recent decades, substantial advancements have been made in elucidating the regulatory mechanisms of IRF family members and their diverse roles in various diseases. A majority of these members are crucial for cellular development, immunity, inflammation, and oncogenesis, serving as in human diseases.32,33,34 However, IRFs may exhibit distinct regulatory effects depending on cell type and environmental context, rendering their roles complex and at times paradoxical and double-edged swords in human health. This review primarily synthesizes the structural characteristics, post-translational modification sites, biological roles, and associated signaling pathways of the IRF family, alongside an exploration of diseases linked to these genes and proteins, with a focus on infections, inflammatory conditions, and a spectrum of cancers, encompassing but not limited to cardiovascular, pulmonary, urinary, reproductive, and skin systems. This review provides a comprehensive insight into the IRF family, underscoring the significance of IRFs as emerging biomarkers and potential therapeutic targets in the realm of human diseases.
IRF protein family: genes, proteins and structures
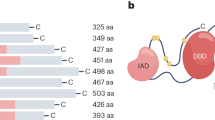
The detailed genes and proteins of IRF family members are presented in Fig. 1. The IRF family encompasses a cohort of transcription factors integral to the regulation of IFNs and the orchestration of immune response. IRF proteins regulate the expression of target genes through their interaction with ISREs or IFN regulatory elements within the DNA. All IRFs feature a highly conserved N-terminal DNA-binding ___domain (DBD), comprising around 120 amino acids that constitute a helix-turn-helix motif. This motif is essential for the identification of specific DNA sequence elements (A/GNGAAANNGAAACT), known as ISRE, present in the promoters of genes for IFN-I, IFN-III, and IFN-stimulated genes (ISGs).27 The C-terminal regions of IRFs contain a conserved ___domain known as IFN association domain1 (IAD1)(IRF3, IRF4, IRF5, IRF6, IRF7, IRF8, and IRF9) or IAD2(IRF1 and IRF2) that have low sequence homology and serve as association domains by which IRFs interact with other IRF members or transcription factors and/or cofactors.1 Many IRF family proteins also contain a regulatory region between the DBD and IAD, which includes multiple phosphorylation sites.

The genes and proteins structures of IRF family members. a The genomic structures. The boxes mark the exons, including non-coding exons (orange) and coding exons (green). blue lines mark the introns. Start and stop codons are indicated. The numbers below the genes indicate the sizes of the exons and introns. b The structures of IRF family proteins. DBD represents the DNA-binding ___domain, LK represents the linker region, IAD represents interferon association ___domain, and AR represents auto-inhibitory region. Active domains (AD) are also marked on the figure. The upper numbers refer to the starting amino acid sites of diferent domains, and the different post-translational modifications of the IRFs are presented, including Phosphorylation, Ubiquitylation, SUMOylation, Methylation, and Acetylation
IRF1 and IRF2
The human IRF1 gene maps to chromosome 5q31.1, spanning 9,165 base pairs (bp) and comprising 10 exons and 9 introns. Human IRF1 protein has an approximate molecular weight of 45 kDa and is composed of 325 amino acids. The IRF1 gene is constitutively expressed in human cells at low basal levels, but can be triggered by diverse stimuli, such as IFNs, tumor necrosis factor (TNF), and interleukin-1 (IL-1).35 The human IRF2 gene is located on chromosome 4q35.1 with 86,822 bp, and contains 9 exons and 8 introns. Through alternative splicing, IRF2 produces two splice variants. Currently, the primary focus of research has been on isoform 1 of IRF2, the full-length variant, which encodes 349 amino acids. IRF2 is constitutively expressed in multiple cell types and its expression can be further induced by viruses and IFN. Despite sharing a similar DBD with IRF1, IRF2 frequently serves an antagonistic role in immune regulation by inhibiting the activation of genes such as IFN-β. This inhibition is achieved through competitive binding to shared sites, thereby functioning predominantly as a suppressor of IRF1-mediated gene activation.
IRF3 and IRF7
The human IRF3 gene is situated on chromosome 19q13.33, spans 6286 bp, and contains 8 exons and 7 introns. A variety of splicing variants of IRF3 exist, with a minimum of five distinct variants identified thus far. The most extensively investigated variant of IRF3 is isoform 1, which is approximately 50 kDa and encodes 427 amino acids. IRF3 is constitutively expressed in many tissues, and the relative steady-state levels of IRF3 mRNA do not increase by virus infection or IFN treatment in cells.21 Situated on chromosome 11p15.5, the human IRF7 gene spans 1923 bp and contains 11 exons and 10 introns. The human IRF7 gene encodes four alternative splicing variants, named IRF7A, IRF7B, IRF7C, and IRF7H. 31,36 Among these, the variant that has been most thoroughly investigated is IRF7A, which is composed of 503 amino acids and has a molecular weight of 55kD. The C-terminal region of IRF7 contains multiple regulatory domains for its activation.37 IRF7, sharing a close relationship with IRF3, operates as a transcription factor that requires activation. The virus-activated ___domain in human IRF7A is vital for the activation of IRF7.38 IRF7 is inherently present in the cytoplasm, with its expression predominantly observed in B cells, plasmacytoid dendritic cells (pDCs), and monocytes in spleen, thymus, and peripheral blood leukocytes.31,36 Various stimuli, including Lipopolysaccharide (LPS), IFN-α, EBV-latent membrane protein 1 (EBV-LMP1), viral infections, and certain chemical agents like phorbol myristate acetate (PMA) and sodium butyrate, can significantly induce the expression of IRF7 in specific cell lines.36,39,40 Additionally, DNA damage agents, virus infection, and EBV-LMP1 can activate IRF7 phosphorylation and nuclear translocation.38,41,42
IRF4 and IRF8
The human IRF4 gene maps to chromosome 6p25.3 with 19,692 bp, contains 9 exons and 8 introns, and gives rise to two splicing variants. The predominant isoform of IRF4, the full-length variant, encodes a protein of approximately 51 kDa, containing 451 amino acids. IRF4 is uniquely expressed within immune system cells and is responsive to various mitogenic stimuli, including PMA/Ionomycin and T-cell receptor (TCR) cross-linking.22,24,25,43 IRF4’s role as a transcriptional activator or suppressor is determined by its interactions with various transcription factors or the DBD on distinct promoters.44,45 The human IRF8 gene is situated on a 23 kb segment of chromosome 16q24.1, and contains 9 exons and 8 introns. IRF8 protein encodes 426 amino acids. IRF8 is predominantly constitutively expressed in lymphoid and myeloid cell lines and induced by IFN-γ, but also in epithelial cells of the intestine, skin, lung, liver, ocular lens, cornea, and heart.46,47,48 IRF8 is highly expressed in both progenitor and mature cells within the B cell, conventional DC1 (cDC1), and pDC lineages, playing a crucial role in their development and functionality.
IRF5 and IRF6
Situated on chromosome 7q32.1, the human IRF5 gene spans 13,007 bp, and comprises 9 exons and 8 introns. Numerous splice variants of IRF5 have been identified, with at least six distinct variants documented thus far. The size of the encoded IRF5 protein varies depending on the splice variant. One of the most extensively studied isoforms of IRF5 is the V1, which encodes a protein approximately 57 kDa and consists of about 498 amino acids. Initially discovered as a key regulator of IFN-I gene expression.28 IRF5 is primarily found in B cells, monocytes, macrophages, and pDCs. During viral-triggered activation of IFN-I genes, there are multiple spliced isoforms of IRF5, each characterized by different patterns of expression specific to cell types, cellular localization, differential regulatory mechanisms, and divergent functional roles.49 The human IRF6 gene maps to chromosome 1q32.2 with 20,526 bp and contains 9 exons and 8 introns. Likewise, IRF6 can produce two primary splice variants (variant 1 and variant 2) via alternative splicing. The existence of these splice variants enables IRF6 to perform distinct roles across diverse cell types and under various physiological conditions. The encoded human IRF6 protein is typically around 56 kDa and consists of approximately 467 amino acids. IRF6 exhibits prominent cytoplasmic expression in various cell types and is crucial for embryonic development, particularly in the epidermal and oral mucosa formation.
IRF9
The human IRF9 gene is situated on chromosome 14q12 with 5301 bp, and contains 9 exons and 8 introns. Human IRF9 protein encodes 393 amino acids. IRF9 is ubiquitously expressed in various cell types and has been identified as playing an essential role in the antiviral defense mechanism mediated by IFN-α/β and IFN-γ.50
Post-translational modifications (PTMs) of the IRFs
PTMs are covalent alterations that modify the properties of a protein through adding a modifying chemical group or peptide moieties to one or several of its amino acid residues.51 A singular protein may undergo modifications by multiple PTM types or be recurrently modified by the same PTM at distinct residues. Key PTMs influencing IRFs encompass phosphorylation, ubiquitination, SUMOylation, methylation, and acetylation, each playing a pivotal role in dictating the functional dynamics, proteostasis, and conformational integrity of these transcription factors (Fig. 1b).
Phosphorylation
IRFs undergo various PTMs, among which phosphorylation stands out as the most critical. Glycogen synthase kinase 3β mediates dual phosphorylation of IRF1 at Thr181 and Ser185, which is required for the regulation of IRF1 turnover by K48-linked polyubiquitination proteasomal degradation.52 Moreover, IκB kinaseε (IKKε) mediates the phosphorylation of the C-terminal region of IRF1, significantly diminishing the stability of IRF1, accelerating it, and inhibiting the transcriptional activity of IRF1.53 In addition, IRF1 undergoes phosphorylated at Tyr109 within the DBD.54,55
Phosphorylation is a crucial PTM of IRF3, as it induces cytoplasm-to-nucleus translocation of phosphorylated IRF3, and stimulates DNA binding and transcriptional activity.56 The two kinases, IKKε and TANK -binding kinase 1 (TBK1) are the major phosphorylation kinases of IRF3.57,58,59 IRF3 features two significant phosphorylation sites: site 1 includes Ser385 and Ser386, whereas site 2 includes Ser396, Ser398, Ser402, Thr404, and Ser405 within the C-terminal region.56,60,61 Moreover, c-Jun N-terminal kinase (JNK) can stimulate the phosphorylation of IRF3 at Ser173in the N-terminal.62 In addition, IFN-I-induced long noncoding RNA-ISIR binds IRF3 at DBD promoting its phosphorylation, dimerization, and nuclear translocation upon infection, consequently facilitating IRF3 activation.63 Recently, Wang et al. found that serine/threonine-protein kinase 38-like phosphorylates IRF3 at Ser303, which prevents IRF3 degradation mediated by the proteasome in the rest state.64 However, mammalian sterile 20-like kinase 1-mediated IRF3 phosphorylation at Thr75 and Thr253 severely disrupted the ability of activated IRF3 to form homodimerization that impairs its transcriptional responses.65
The phosphorylation sites of IRF4 include Tyr121 and Tyr124. LMP1 promotes IRF4 tyrosine phosphorylation and significantly stimulates its transcriptional activity.66 Moreover, Rho-associated coiled-coil-containing protein kinase 2 can phosphorylate IRF4 to regulate the production of IL-17 and IL-21.67 In microglia, IL-1 receptor associated kinase 4 (IRAK4) phosphorylates both IRF4 and IRF5 by forming a Myddosome with myeloid differentiation primary-response protein 88 (MyD88)/IRF5/IRF4.68
The phosphorylation sits of IRF5 include Ser158, Ser309, Ser451, Ser466, and Ser462.69,70 In myeloid cells, phosphorylation at Ser462, facilitated by IKKβ, activates IRF5, which triggers IRF5 dimerization of and subsequent nucleus translocation.71 However, IKKα-induced phosphorylation of IRF5 has an inhibitory effect on the transcriptional activation of IFN-I and the promoters of inflammatory cytokines.72
The phosphorylation of IRF6 at Ser413 and Ser424 primes IRF6 for activation.73 In addition, receptor interacting protein kinase 4 (RIPK4) directly regulates the trans-activator activity and nuclear translocation of IRF6 via phosphorylating its C-terminal ___domain at Ser413 and Ser424.74
The kinases implicated in the phosphorylation of IRF7 include IKKε, IKKα, IRAK1, and TBK1.57,75,76,77 In response to viral infection, IRF7 is phosphorylated at Ser477 and Ser479 and activated by TBK1 and IKKε.38 Additionally, phosphorylation at Ser471, Ser472, Ser483, Ser484, and Ser487 also contributes to the activation of IRF7. However, the exogenous expression of protein phosphatase 1(PP1) subunits, heat shock protein 70, vaccinia virus E3L protein, and the open reading frame 45 of Kaposi’s sarcoma-associated herpesvirus inhibit IKKε-stimulated IRF7 phosphorylation and significantly reduce IRF7 transcriptional activity.78,79,80,81
Ubiquitylation
Ubiquitination is a universal reversible PTM that can either activate or deactivate protein, with IRFs being tightly regulated by ubiquitination in various respects.82 Ubiquitination can either positively or negatively influence the stability, activation, and transcriptional activity of IRFs. The fundamental element of ubiquitination is ubiquitin, which is covalently attached to one or more lysine residues in cellular involving three classes of enzymes.83 Ubiquitin itself contains seven lysine residues and one N-terminal methionine residue. Each of these can be further conjugated with another ubiquitin to form ubiquitin chains of different linkages.
IRF1 is characterized by a brief, undergoing rapid degradation through the ubiquitin-proteasome pathway.84 K63-linked ubiquitination of IRF1 contributes to its activation, while K48-linked ubiquitination contributes to its degradation.85 Notably, HIV-1 viruses have developed strategies to exploit this ubiquitin-proteasome pathway to inactivate IRF1 function and evade the host immune defense mediated by IRF1.86 IRF1 K63-linked ubiquitination is mediated by TNFR-associated factor 6 (TRAF6) and cellular inhibitor of apoptosis 2 (clAP2).87 In response to IL-1 stimulation, clAP2 mediates the K63-linked polyubiquitination of newly synthesized IRF1, leading to its activation.88
MGF360-14L, a viral non-structural protein, facilitates the degradation of IRF3 via tripartite motif-containing protein (TRIM) 21-mediated K63-linked ubiquitination of IRF3.89 Cellular E3 ligases c-Cbl, RTA-associated ubiquitin ligase (RAUL), RBCC protein interacting with PKC1 (RBCK1), and Midline-1 (MID1) target IRF3 for K48-linked polyubiquitination, leading to its proteasome-dependent degradation. Viral infection leads to the induction of RBCK1, which subsequently catalyzes the ubiquitination and degradation of IRF3.90 During viral infection, MID1 inhibits IFN-I production by interacting with IRF3 and negatively regulating IRF3 protein levels.91 MID1 induces the ubiquitination of IRF3 at Lys313 playing a role in the cellular antiviral response, which is governed by a negative feedback mechanism. Jumonji ___domain-containing protein 6 promotes activated IRF3 K48 ubiquitination and degradation by ring finger protein 5.92 Sentrin/SUMO-specific protease 2 catalyzes K48-linked ubiquitination of IRF3 at Lys87 and competitively promotes IRF3 deSUMOylation at Lys70.93
IRF3 ubiquitination not only leads to its degradation but also contributes to its activation. Specifically, the activation of IRF3 in the RLR-induced IRF-3-mediated pathway of apoptosis (RIPA) requires linear polyubiquitination of two specific lysine residues on IRF3 by the linear polyubiquitinating enzyme complex.94 However, this pathway can be inhibited by Otulin, a deubiquitinase, which removes linear polyubiquitin chains, resulting in IRF3 deubiquitinating.95
Moreover, E3 ligase ring finger protein 2 (RNF2) promotes the ubiquitination and degradation of IRF4 in colon cancer.96 MiR-155-5p contributes to the development of childhood acute lymphoblastic leukemia (ALL) by the Casitas B-lineage lymphoma (CBL)-mediated degradation of IRF4 via ubiquitination.97 In addition, CBLs exhibit elevated expression levels in germinal center light zone B cells, where they promote the ubiquitination and degradation of IRF4.98 However, the ubiquitin specific peptidase 4 (USP4) interacts with and deubiquitinate IRF4 to stabilize IRF4 protein, thereby promoting IRF4 function to facilitate IL-4 expression in Th2.99
K63-linked ubiquitination of IRF5 contributes to its activation and increases its nuclear translocation.100 TARF6 and Pellino-1 have been identified as the ubiquitin E3 ligases for IRF5, which target and promote K63-linked ubiquitination of IRF5. In human and mouse M1 macrophages, the interaction between Pellino-1 and IRF5 in the cytoplasm activates IRF5 and increases its nuclear translocation via K63-linked ubiquitination.101,102
The RTA immediate-early nuclear transcription factor, which is encoded by Kaposi’s sarcoma-associated herpesvirus, facilitates the ubiquitination and degradation of the IRF7 protein in a proteasome-dependent.103 Additionally, RAUL regulates IFN-I via targeting both IRF7 and IRF3 for K48-linked polyubiquitination and proteolysis.104 LMP1-induced antiapoptotic factor A20 possesses both deubiquitinase and ubiquitin E3 ligase activities, and negatively regulates LMP1-stimulated IRF7 K63-liked ubiquitination and activity during EBV latency.105 N-Myc and STATs interactor is a Sendai virus-inducible protein, which promotes the IRF7 K48-linked ubiquitination and proteasome-dependent degradation.106 Similarly, TRIM35 also promotes the K48-linked ubiquitination of IRF7 and induces its degradation via a proteasome-dependent pathway. The interaction of Fas-associated death ___domain (FADD) with TRIM21 can enhance TRIM21’s ubiquitin ligase activity, both of them can directly ubiquitinate IRF7, affect its phosphorylation status, and interfere with TRAF6 ubiquitin ligase activity.107
Additionally, IRF7 can be activated by the EBV-LMP1 via receptor interacting protein (RIP)-dependent K63-linked ubiquitination.108 Further studies found that both TRAF6 and its E3 ligase activity are required for LMP1-stimulated IRF7 ubiquitination. IRF7 is ubiquitinated by TRAF6 at multiple sites, but the K63-linked ubiquitination sites are independent of its C-terminal functional phosphorylation sites.109 Nevertheless, TAR RNA binding protein 2, an inhibitor of IRF7, inhibits the K63-linked ubiquitination and phosphorylation of IRF7.110
CBL also mediates IRF8 ubiquitination, leading to the degradation of IRF8.111 Ro52 (also called TRIM21) can interact with and ubiquitinate IRF8 in a non-degradation pathway. This interaction in turn enhances IL-12p40 expression in an IRF8-dependent manner.112 Similarly, USP4 also stabilizes IRF8 protein levels in regulatory T cells (Tregs) by interacting with IRF8 via a K48-linked deubiquitinase.113 Likewise, IRF9 can be ubiquitinated and degraded by herpes simplex virus (HSV) type 2 ICP22 protein, which functions as a novel E3 ubiquitin protein ligase.114
SUMOylation
SUMOylation, is a critical PTM that is catalyzed by a limited set of modifying enzymes yet dynamically regulates a vast array of target proteins. Small ubiquitin-like modifiers (SUMOs) are members of the ubiquitin-like family of proteins that predominantly target nuclear proteins.115 Five SUMO family members have been identified in mammals.116 SUMOylation is pivotal in the regulation of nuclear processes, including transcription, nuclear body formation, nucleocytoplasmic transport, RNA processing, cell cycle progression, DNA repair, chromosomal functions, and signal transduction.117,118 Similar to ubiquitylation, SUMOylation is reversible, as SUMO proteases can remove SUMOs from their substrates.118,119 IRFs are also regulated by SUMOylation
Lys275 is identified as the primary SUMOylation site of IRF1. The protein inhibitor of activated STAT3 serves as both a SUMO-1 ligase and an inhibitor of IRF1’s transcriptional functions.120 Additionally, the SUMO-conjugating enzyme Ubc9 suppresses IRF1’s transcriptional activation by inducing IRF1 SUMOylation.121 Moreover, some studies have demonstrated that SUMOylated IRF1 may act as an oncogenic protein in tumor cells.122 SUMOylated IRF1 is elevated in tumors, which inactivates its tumor suppressor function by repression of its transcriptional activity that facilitates resistance to the immune response.123 Furthermore, treatment with alpha-lipoic acid induced IRF1 SUMOylation by increased SUMO-1 in an IL-1β-stimulated chondrocyte model.124 The level of SUMOylated IRF1 was significantly elevated in the myocardial infarction (MI) group treated with 5-azacytidine.125
IRF2 SUMOylation sites include Lys137, Lys293, and Lys166. SUMOylation of IRF2, catalyzed by SUMO-E3 ligase PIASy, represses its transcriptional activity in an histone deacetylase (HDAC)-dependent manner.126 SUMOylation of IRF2 has minimal effects on its nuclear localization and DNA-binding activity. However, it enhances the inhibition of IRF1’s transcriptional activity while reducing the capacity to activate ISRE and H4 promoters.
During vesicular stomatitis virus infection, both IRF3 and IRF7 undergo phosphorylation as well as modification by SUMO1, SUMO2, and SUMO3.127 The SUMOylation of IRF3 at Lys152 and IRF7 at Lys 406 is independent of their phosphorylation, and vice versa. However, some studies have found that SUMOylation of IRF3 leads to a reduction in IRF3 phosphorylation and IFN synthesis.128 SUMOylation of IRF3 and IRF7 negatively regulates IFN transcription.127 The Ebola Zaire virus VP35 protein inhibited IFN transcription in DCs by increasing PIAS1-mediated SUMOylation of IRF7.129 The SUMO conjugation sites of IRF7 include Lys406 and Lys452.127,130 The EBV-LMP1 can limit the capacity of IRF7 to initiate innate immune responses via promoting IRF7 SUMOylation at Lys452.130 The TRIM28 is a specific SUMO E3 ligase and a negative regulator of IRF7.131
In addition, Ubc9-mediated IRF4 SUMOylation enhanced its nuclear localization and stability.132 Similarly, IRF5 and IRF8 can undergo SUMOylated.133 However, the SUMOylation of IRF8, catalyzed by SUMO3 at the Lys310, can be reversed by SUMO-specific protease (SENP) 1 and SENP3.134,135
Methylation and acetylation
Protein methylation refers to the transfer of methyl to a protein residue. Acetylation modification is a reversible and evolutionarily conserved PTM. Some IRFs (IRF1, IRF2, IRF3, IRF7, and IRF9) also undergo methylation or acetylation.
In U937 cells treated with PMA, both IRF1 and IRF2 undergo acetylation facilitated by p300 and p300/ CREB-binding protein (CBP)-associated factor (PCAF).136 The p300-mediated IRF1 acetylation sites include the N-terminal Lys39 and Lys78.137 In addition, IRF1 can be specifically acetylated by KAT8 at Lys78.138 The acetylation sites of IRF2 include N-terminal DBD Lys75 and Lys78. Furthermore, some studies have shown that the acetylation of IRF2 is dependent on cell growth.139,140 The monomethylation of IRF3 at Lys366 by nuclear receptor-binding SET ___domain 3 (NSD3), enhances the transcriptional activity of IRF3 in antiviral innate immunity.141 This is because NSD3-mediated IRF3 methylation obstructs IRF3 dephosphorylation by disrupting PP1’s association with IRF3, thereby maintaining IRF3 phosphorylation.142 Furthermore, LPS induces the arginine methylation of IRF3 at Arg285, leading to its dimerization and promoting its translocation from the cytoplasm to the nucleus.143
KAT8 mediates IRF3 acetylation at Lys359 through its MYST ___domain, which leads to the inhibition of IRF3 recruitment to IFN-I gene promoters and a decrease in its transcriptional activity.144 In vivo, IRF7 undergoes acetylation at Lys92 located in the DBD by the histone acetyltransferases PCAF and GCN5, resulting in impaired DNA binding capability.145 A subsequent study has shown that sirtuin1 (SIRT1) -mediated DBD deacetylation is a pivotal mechanism in the activation of IRF3 and IRF7.146 Upon viral stimulation, viral interferon regulatory factor 3 engaged liquid-liquid phase separation with ISRE DNA and compartmentalized IRF7 in the nucleus, thus stimulating the expression of IFN-I. In addition, IRF5 and IRF9 also undergo lysine acetylation.147,148
Biological functions of IRFs
Regulation of immune cell development
Diverse studies have shown that the IRF family can regulate immune cell differentiation. Herein, IRF family members with their functions and molecular mechanisms in immune cells are discussed in detail. The subtypes of myeloid cells and lymphoid cells are pesented in Fig. 2a.

The regulatory effects and molecular mechanisms of IRFs on immune cell development. a The subtypes of myeloid cells and lymphoid cells. b The induction effect of IRFs on DC maturation and cytokine production. c The role of IRFs in myeloid differentiation and MDSC aggregation. d Regulation of IRFs in Natural Killer Cells development. e Multistep regulation of B cells and plasma cells by IRFs. f IRFs regulate T cell development and differentiation
The induction effect of IRFs on DC maturation and cytokines production
DCs constitute a specialized subset of hematopoietic cells endowed with the capacity to secrete IFN-I and a plethora of other cytokines. As the quintessential antigen-presenting cells (APCs), DCs are instrumental in initiating both innate and adaptive immune responses.149 Characterized by their high expression of MHC molecules, DCs, upon detecting invading pathogens via pattern recognition receptors (PRRs), secrete various cytokines and present antigen-MHC complexes to T cells, thereby eliciting helper T cell (Th) responses or inducing immunological tolerance.150
DCs constitute a heterogeneous population, comprising subpopulations with distinct functionalities, which can be categorized into conventional DCs (cDCs) and pDCs based on unique surface markers. In mice, spleen DCs are further subdivided into at least four subgroups: CD4+ DCs, CD8α+ DCs, CD4-CD8α- (double negative, DN) DCs, and pDCs.150 In humans, cDCs are bifurcated into two subgroups: CD141+cDC1 and CD1c+DC2 cells, with their murine counterparts being CD11b-CD103+ (CD8α+) cDC1 and CD11b+cDC2. These cell populations, characterized by different gene expression profiles, perform distinct functions. pDCs are prolific producers of IFN-I, whereas cDCs generate both pro-inflammatory and anti-inflammatory cytokines, such as IL-10 and IL-12. cDC1 cells are pivotal for eliciting CD8+T cell responses under viral or tumoral challenges, while cDC2 cells exhibit a robust capacity to initiate CD4+T cell responses.151
The IRF family proteins governs the differentiation and functional activity of DCs (Fig. 2b). IRF4 and IRF8 are selectively instrumental in the development of specific DC subgroups.152,153 These two IRFs share overlapping roles in driving the general process of DC development, yet they also possess distinct activities that stimulate the expression of subgroup-specific genes, resulting in the emergence of functionally diverse DC subpopulations. During DC development, DN DCs appear to represent the prototypical DC subset, differentiating into various DC subtypes and functions under the differential regulation of IRFs. IRF4 is crucial for producing CD4+DCs, while CD8α+ DCs require IRF8. Both of the two IRFs are instrumental in the development of CD4-CD8α- DCs and pDCs.154 These cells express both IRF4 and IRF8, with CD8α+ DCs expressing high levels of IRF8 and CD4+DCs predominantly expressing IRF4.154 In Irf8-/- mice, there is a notable absence of CD8α+ DCs and pDCs, whereas in Irf4-/- mice, the numbers of CD4+ DCs are significantly diminished.152,155,156 Mice deficient in both IRF8 and IRF4 retain only a minor population of CD4-CD8α- DCs, completely lacking other DC subtypes in the spleen.154
Without IRF8, committed cDC1 cells acquire transcriptional, functional, and chromatin accessibility characteristics reminiscent of cDC2 cells. While IRF8 is not essential for the survival of committed cDC1 cells, it is critical for preventing their transdifferentiation into cDC2-like cells.157 Furthermore, IRF1 and IRF2 also modulate the development of DC subgroups.158,159 In Irf1-/- mice, pDCs predominate, while the cDC population, particularly the CD8α+ subset, is selectively diminished.158 The capacity of IRF1-deficient spleen DCs to produce pro-inflammatory cytokines such as IL-12 is significantly compromised. In Irf2-/- mice, spleen CD4+CD11b+DCs exhibit pronounced selective deficits.159
IRF proteins also regulate the induction of IFN-I in DCs.160 Among them, IRF3 and IRF7 are widely recognized as crucial transcription factors for IFN induction.32,161 IRF7, activated under toll like receptor (TLR) signaling, promotes IFN induction in pDCs, cDCs, and non-DC cells.160,162,163,164 IRF3 primarily facilitates IFN induction in fibroblasts and is not necessary for IFN induction in DCs.151 IRF3 is the primary early regulatory factor that induces IFN-I during intracellular viral infection.165 While IRF5 is not required for IFN induction, it can enhance the production of non-IFN pro-inflammatory cytokines through distinct mechanisms.160,163,166 However, surprising findings from in vivo studies suggest the existence of an alternative IFN induction pathway that operates independently of IRF3, IRF5, and IRF7. It was expected that IRF3-5-7 triple knockout mice would exhibit impaired IFN-I production, yet IFN-I activity was still detected in their serum.167 The most likely candidate responsible for this induction appears to be IRF1. IRF1 is expressed widely and can enhance early IFN production by modulating the phosphorylation and localization of IRF3.168 It may also compensate for the role of IRF7 as a positive regulator of IFN, establishing a positive feedback mechanism to sustain IFN production.32 The generation of IFN-I in DCs is heavily reliant on IRF8, particularly during the feedback phase of IFN gene induction. Exogenous expression of IRF8 can rescue the development of pDCs and CD8α+ DCs in vitro, triggering IFN-I and IL-12p40 production, whereas IRF4 does not exert the same effect.169 Conversely, the introduction of IRF4 can restore the expression of selectively expressed genes in CD4+DCs. The activation of IFN-I in pDCs is mediated by a MyD88-dependent signaling pathway, reliant on TLR3 and retinoic acid inducible gene I (RIG-I)-like receptors(RLRs).160 The cytokines produced by activated DCs, including IFN, subsequently promote DC maturation and alter DC phenotypes and functions, with IRF family proteins potentially involved in regulating these processes.
Beyond IFN-I, IRF family proteins are also pivotal in the induction of other pro-inflammatory cytokines. IL-12p70, produced by APCs and B cells, is a heterodimeric pro-inflammatory cytokine secreted into the extracellular milieu.170 IL-12 can induce IFN-γ production. IL-12p70 comprises two subunits, p40 and p35. IL-12p40 is essential for inducing IFN in Natural Killer (NK) and T cells, with its production in DC cells being dependent on IRF8.171 IRF8’s role in inducing IL-12 extends beyond regulating DC cell development, it also directly participates as a transcriptional activator in the gene transcription regulation of these cytokines.1 IRF1 is involved in the induction of IL-12p40 in DC cells and is necessary for macrophages to fully induce IL-12p40.27,160,172 Additionally, IRF5 is involved in activities that include the induction of pro-inflammatory factors such as IL-12p40, TNF-α, and IL-6. This process is mediated by TLR signaling, which stimulates IRF5’s translocation to the nucleus, interaction with MyD88 and TRAF6, and initiation of IL-12p40 expression.151
In conclusion, the IRF protein family is integral to the development of DCs, and the induction of IFN-I within these cells. The production of IFN in response to pathogen infection is crucial for activating effective innate immunity to combat infection before the establishment of adaptive immunity. IRFs endow DCs with the necessary versatility for the optimal regulation of immune responses.
The role of IRFs in myeloid differentiation and myeloid derived inhibitory cells (MDSCs) aggregation
The differentiation of macrophages and granulocytes is regulated by IRFs
The Fig. 2c reveals the role of IRFs in macrophages and granulocytes differentiation. IRF8 is predominantly localized within hematopoietic cells and is variably expressed throughout the differentiation, proliferation, and apoptotic processes during myeloid cell development, where it orchestrates their core functions.169 The differentiation of myeloid progenitor cells yields granulocytes and macrophages,173 with studies on Irf8-/- myeloid progenitors revealing that IRF8 directs the trajectory of differentiation, favoring macrophage formation while suppressing granulocyte differentiation. Irf8-/- mice manifest symptoms akin to human chronic myeloid leukemia (CML).174 Irf8 gene deletion leads to an increased progenitor cell population that is hypersensitive to granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF). These progenitors exhibit a diminished response to macrophage colony-stimulating factor (M-CSF) but retain the capacity to differentiate into granulocytes in the presence of M-CSF.175 IRF8 expression is sustained in macrophages but reduced in granulocytes. It governs several pivotal genes implicated in myeloid cell proliferation and apoptosis, inhibiting cell growth and promoting apoptosis.175,176 The absence of IRF8 disrupts the myeloid differentiation program, skewing differentiation towards granulocytes and culminating in a CML-like syndrome. Notably, a marked reduction in IRF8 transcription levels has also been noted in cells derived from human CML patients.177
Mammalian rapamycin target (mTOR) affects M-CSF receptor CD115 expression by regulating the STAT5-IRF8 axis, controlling the development of monocytes/macrophages in the early stages.178 Upon pathogenic stimulation of human blood monocytes, a transcription factor complex comprising IRF8 and PU.1 associate with IFN-β to initiate Ets-IRF complex element (EICE) binding in subregions, recruit IRF3, and swiftly induce IFN-β expression, quickly initiate innate immune response to pathogens.179 IRF8 also upregulates multiple genes essential to macrophage function, including those linked to endosomes and lysosomal enzymes (e.g., cathepsin C, lysozyme, cystatin C), as well as genes that stimulate macrophage adhesion and migration (such as the Dab2 gene).180,181 IRF8 can form heterogeneous complexes with other transcription factors, such as the ETS family member PU.1 and IRF1, as a co-activator of various IFN-induced genes in macrophages, indirectly modulating the transcription of IFN-γ-responsive genes at the gamma-activated site (GAS), including those encoding IL-12p40, IL-12p35, gp91phox, p67phox, TLR4, TLR9, inducible nitric oxide synthase (iNOS), Fcγ receptor I (FcγRI), IRF8 itself, IL-18, chemokine ligand 5 (CCL5)/RANTES, and the phagolysosomal natural resistance-associated macrophage protein 1 (NRAMP1), thereby bolstering host defenses against pathogens.27,169,182,183 IRF8 regulates the nucleotide-binding ___domain, leucine-rich repeat-containing receptor (NLR) family of apoptosis inhibitory proteins, and nucleotide-binding oligomerization ___domain-containing (NOD)-like receptor family caspase activation and recruitment domains (CARD) containing 4 (NLRC4) inflammasome activation, eliciting cellular responses to bacterial proteins that lead to caspase-1 activation and subsequent secretion of pro-inflammatory cytokines IL-1β and IL-18, which promote pyroptosis and intracellular pathogen resistance within macrophages.184 Additionally, IRF8 can bind to the promoters of autophagy-related genes, encompassing all stages of autophagy in macrophages under conditions such as IFN-γ/TLR stimulation or pathogen infection.185 Macrophages possess intrinsic mechanisms to fine-tune IRF8 transcriptional activity, including the deSUMOylation of IRF8 by SUMOs, ubiquitination by ubiquitin ligase TRIM21, and nucleosome remodeling by IRF8, all of which influence the transcriptional output of IRF8 target genes.135,186
IRF1 and IRF2 play critical roles in the regulation of myeloid cell differentiation. Myeloid cells deficient in IRF1 exhibit impaired maturation. In macrophages stimulated by IFN, the IFN-inducible transcriptional activator IRF1 targets and modulates the expression of a multitude of genes that contain ISREs. These genes include those encoding guanylate-binding proteins, iNOS, caspase-1, cyclooxygenase-2 (Cox-2), class II transactivator (CIITA), and gp91phox.1,187,188,189 Under inflammatory conditions, IRF1 can also form complexes with IRF8, co-activating IFN-induced gene expression. Conversely, a deficiency in IRF2 results in the expansion of eosinophil populations, leading to elevated IL-4 expression and the promotion of Th2 cell polarization. Macrophages lacking IRF2 (Irf2-/-) show increased expression of caspase-1, suggesting that IRF1 and IRF2 have opposing regulatory effects on caspase-1 expression. Despite this, IRF1 and IRF2 demonstrate synergistic effects in the regulation of IL-12p40 and Cox-2 expression.1
Regulatory functions of IRFs in microglia and osteoclasts
Macrophages are distributed across various tissues and organs, with their classification—including microglia, osteoclasts, and alveolar macrophages (AMs)-based on ___location and function.190,191 Microglia, the resident macrophages of the central nervous system, are integral to the glial system, contributing to neural homeostasis and protection.192,193 The Fig. 2c reveals the role of IRFs in microglia differentiation. Within microglia, IRF8 establishes a positive feedback loop with the transcription factor PU.1, sustaining IRF8 expression.194 Following neural injury, IRF8 activates target genes that transition microglia to reactive phenotypes, which can have protective or neurotoxic effects on the nervous system.195,196 In reactive microglia, IRF1 collaborates with IRF8 to initiate transcription of IL-1β.197 The absence of Irf8 in microglia can lead to an increase in TNF-α, mediate an increase in hippocampal nerve excitability, and drive fatal epilepsy.198
Studies have identified IRF7 as a factor that promotes glioblastoma multiforme progression. IRF7 regulates the expression of inflammatory cytokines, polarizes microglia towards an M2 phenotype, fosters an immunosuppressive tumor microenvironment, and facilitates tumor growth, invasion, and immune evasion.164
Osteoclasts, specialized terminal differentiation cells within the monocyte-macrophage lineage, fuse with monocytes to form multinucleated giant cells involved in bone resorption.199 The Fig. 2c presents the role of IRFs in osteoclast differentiation. IRF8 acts as a negative regulator in osteoclast differentiation and maturation.200 The absence of IRF8 leads to an increase in osteoclasts, contributing to bone metabolic disorders and playing a pivotal role in osteoclast-related inflammatory diseases such as multiple root resorption.201,202,203,204
The regulatory role of IRFs in MDSCs
MDSCs represent a heterogeneous set of myeloid progenitor cells, including precursors unable to differentiate into macrophages, granulocytes, and DCs.205 These progenitor cells can become arrested at various stages of maturation due to influences such as tumors, inflammation, trauma, or certain autoimmune diseases, resulting in a population with potent immunosuppressive capabilities.206 MDSCs are a hallmark of malignancy and a promising target for cancer treatment.207
The regulatory role of IRFs in MDSCs is shown in the Fig. 2c. IRF8 is a critical factor in myeloid cell development and acts as a negative regulator of MDSCs.208 Within the tumor microenvironment, tumor cells and infiltrating stromal cells release cytokines like G-CSF and GM-CSF, which activate the Janus kinase (JAK)/STAT signaling pathway and suppress IRF8 expression in MDSCs. This suppression of IRF8 hinders T cell activation and infiltration, thereby diminishing the anti-tumor immune response.209 In murine models, overexpression of IRF8 has been shown to restrict MDSC accumulation, mitigate the immunosuppression exerted by MDSCs, counteract their tumor-promoting effects, and improve the effectiveness of immunotherapies.208
IRF8 also interacts with the promoter of the Bax gene, influencing the Bax-mediated apoptotic pathway and promoting apoptosis of MDSCs.210 Additionally, the expression of IRF4 within MDSCs is reduced,211 and IRF4 deficiency contributes to MDSC proliferation in the tumor microenvironment,212 Enhancing IRF4 expression can decrease MDSC numbers and attenuate their immunosuppressive function by inducing differentiation.213 Besides, as a tumor suppressor, IRF7 is known to downregulate the expression of S100A9, a protein that inhibits the expansion and aggregation of granulocytic MDSCs, thereby curtailing tumor cell metastasis and dissemination.214
Regulation of NK Cells development by IRFs
IRF1 exerts a selective impact on the stromal cells within the developmental niche of NK cells (Fig. 2d).215 IRF1 is implicated in regulating the expression of IL-15 in these stromal cells, a cytokine that is essential for NK cell development.1 Consequently, in IRF1 knockout (Irf1-/-) mice, a significant reduction in NK cell populations is observed, accompanied by the abrogation of NK cell functions, including cytotoxicity and IFN-γ secretion, particularly following IL-12 stimulation.
Similarly, the absence of IRF2 also impairs the development and functionality of NK cells (Fig. 2d). However, the underlying mechanism appears to differ from that of IRF1, as it may involve the promotion of accelerated apoptosis in NK cells.216 This suggests that while both IRF1 and IRF2 are integral to NK cell biology, they contribute through distinct pathways, with IRF1 primarily influencing the cytokine milieu necessary for NK cell maturation, and IRF2 affecting NK cell survival. In a study on prostate cancer, researchers found that overexpression of IRF7 can increase IFN-β, significantly enhance NK cell activity, and limit bone metastasis of prostate cancer cells (Fig. 2d).164
Multistep regulation of B cells and plasma cells by IRFs
IRF4 is pivotal in B cell ontogeny, encompassing pre-B cell differentiation, receptor editing, germinal center reactions, and plasma cell formation.217 IRF4 expression is characteristic of immature B cells and is subsequently downregulated as antigen-activated B cells transition into the germinal center. Within the germinal center, IRF4 protein is scarcely detectable in the majority of B cells, including both centripetal and most centroblasts.218 The Fig. 2e shows the regulation of B cell differentiation by IRFs.
Both IRF4 and IRF8 are co-expressed in immature B cells, where they collaboratively modulate B cell differentiation.1 Although their regulatory roles appear to be overlapping, compelling evidence indicates that a block in B cell development occurs exclusively in mice lacking both IRF4 and IRF8, manifesting as a pre-B cell stage arrest.219 In mice deficient in either Irf4 or Irf8 alone, B cell maturation arrest can be compensated for by the normal expression of the other factor. The enhancer of the conventional immunoglobulin light chain gene contains an EICE, where IRF4 and IRF8 interact with ETS transcription factors such as PU.1 or SpiB.217
Upon antigen stimulation, B cells migrate to the germinal center within lymphoid tissues, where IRF4 expression is suppressed and IRF8 expression is augmented in the germinal center’s dark zone.220,221 In the light zone, centrocytes undergo affinity maturation and isotype switching to evade apoptosis and differentiate into plasma cells that secrete high-affinity antibodies. Here, IRF8 expression diminishes while IRF4 expression escalates.222,223 In the dark zone germinal center response, IRF8 critically regulates genes such as activation-induced cytidine deaminase (AICDA or AID) and B cell leukemia/lymphoma 6 (BCL6).224 The AICDA gene encodes AID protein, a crucial enzyme in the processes of DNA strand unwinding and recombination, governing somatic hypermutation and class-switch recombination, leading to diverse and highly specific antibody generation. BCL6 is a transcriptional repressor that orchestrates the germinal center response. IRF4 can regulate the Aicda and PR/SET ___domain 1 (Prdm1) genes, where low levels of IRF4 can stimulate Aicda, facilitating AID function, followed by an upsurge in IRF4 expression. Elevated levels of IRF4 induce Prdm1 expression, which in turn represses Bcl6 and Aicda through transcriptional inhibition, culminating in the maturation and differentiation of plasma cells.222
Thymocyte development and Th1/Th2 differentiation regulated by IRFs
T cells orchestrate cellular immune responses within the body, possessing the ability to directly eliminate target cells or secrete cytokines to amplify immune responses. Additionally, they play a role in modulating B cell-mediated humoral immunity. T cells constitute a diverse and heterogeneous group, with various functional subsets at different stages of development. Based on their roles in immune responses, T cells can be categorized into Th, effector T cells (Te), cytotoxic T lymphocytes (CTLs), Tregs, memory T cells, and inhibitory T cells, among others.225 Dysregulation of T cell subsets is commonly observed in immune pathologies. IRF family members have been implicated in the regulation of T cell development and differentiation (Fig. 2f). Activated naive CD8+ T cells can differentiate into cytotoxic cells, secreting various cytokines and categorized into Tc1 (also known as CTL), Tc2, Tc17, Tc9, and Tc22 subsets.226 Most CD8+ T cells differentiate into Tc1, which is stimulated by IL-12 and IL-2 to secrete cytokines such as IFN-γ and TNF-α, directly targeting and killing cells. Tc9 and Tc17 secrete IL-9 and IL-17, respectively.
IRF4 is significant for the differentiation identity of these CD8+ T cell subsets. Although not necessary for CTL activation and proliferation, IRF4 is crucial for maintaining and functioning their phenotype. Irf4-/- mice display functional defects in effector CTLs, with IRF4 influencing the upstream effector functions of transcription factors T-bet, BLIMP-1 (also known as PRDM1), and the formation of memory CTLs. Tc9 cells, akin to Th9 cells, produce IL-9 and IL-10. The molecular mechanism by which IRF4 affects Tc9 development parallels that in Th9, regulating IL-9 expression via binding to its promoter.227 Tc17 cells express IL-17, and Irf4-/- mice also exhibit defects in Tc17 differentiation.228,229
IRF1 is pivotal in CD8+ T cell development, regulating the expression of genes essential for lineage selection in developing thymic CD8+ T cells (Fig. 2f). It also influences the activation of CTLs.230 Moreover, the activation of CTLs is positively regulated by both IRF4 and IRF8.212 While IRF2 also contributes to CTL activity, its absence does not significantly impair CTL function. Unique among its family, IRF2 is a negative regulator that modulates immune responses, maintaining balance in the IFN-I signaling pathway and preventing CD8+ T cells from overreacting to antigenic stimulation, which could otherwise lead to detrimental effects on the body.231
IRF1 is not necessary for Th1 differentiation of CD4+ T cells; however, its absence can lead to developmental defects in various cell types, ultimately constraining Th1 differentiation.232 Mice deficient in IRF1 are devoid of NK cells, which are critical for secreting IFN-γ that in turn stimulates macrophages to produce IL-12, a cytokine indispensable for Th1 differentiation. Additionally, CD4+ T cells with IRF1 deficiency exhibit a delayed response to IL-12.232 Mice lacking IRF2 also present with differentiation impairments in both NK and Th1 cells, with IRF1 and IRF2 acting together to enhance the expression of IL-12p40. Furthermore, IRF2 serves to restrict the proliferation of eosinophils that secrete the Th2 cytokine IL-4, thereby attenuating Th2 cell polarization.233
IRF4 expression in mature T cells does not influence thymocyte development; however, it does affect cytokine production and the cytotoxic capabilities of T cells, potentially impacting their apoptotic and proliferative capacities.23 IRF4 is indispensable for the differentiation of CD4+ T cells into Th2 cells, while IRF8 exerts an antagonistic regulatory effect.45 The absence of IRF8 leads to a deficiency of macrophages and, as well as a compromised Th1 response due to its essential role in IL-12 gene expression. Together, IRF4 and IRF8 orchestrate the Th1/Th2 balance, influencing the generation of plasma cells that secrete highly specific antibodies.154 They are integral to the induction of both cellular and humoral immunity, underscoring their significance in the immune response.
Th1 cells are integral to the host defense mechanism against viruses, bacteria, and intracellular pathogens, producing cytokines such as GM-CSF, IL-2, TNF-β, and IFN-γ.234 IRF5 is implicated in directing T cell differentiation towards the Th1 lineage, with a reduction in the Th1 subset observed in mice with systemic Irf5 knockout.235 IFN-γ stimulation of CD4+ T cells activates STAT1, leading to its nuclear translocation and then production of the transcription factor T-bet. IRF5 enhances T-bet’s IFN-γ production at the encoding gene locus, mediates chromatin remodeling, and together with STAT4, induces IFN-γ expression, driving Th1 polarization. A deficiency in IRF5 results in decreased efficiency of Th1 polarization initiated by T-bet.236
Th2 cells are involved in the immune response against parasitic infections, allergies, and chronic inflammation.237 The differentiation mechanism of Th2 cells is not completely understood; however, IRF4 is a crucial factor in the development of the Th2 subset and acts as an inhibitor of IRF5 transcription.238,239 IL-4 is a critical cytokine for Th2 polarization. Antigen stimulation leads to the upregulation of IRF4, while IL-4 induces the phosphorylation and nuclear translocation of STAT6. Phosphorylated STAT6, in conjunction with IRF4, activates the transcription factor GATA binding protein 3 (GATA3). Transcription factors IKAROS family zinc finger 1 (Ikaros or IKZF1) and GATA3 can promote the transcription of Th2 polarization cytokines IL-4, IL-5, and IL-13.240,241,242 Within these regulatory mechanisms, IRF5 negatively regulates Ikaros transcription, whereas IRF4 antagonizes the MyD88-IRF5 interaction, inhibiting IRF5 activation.243 A positive feedback loop exists between IRF4 and GATA3, and together with Ikaros, they can suppress the Th1 transcriptional network, ultimately reinforcing the Th2 phenotype.
Th9 cells, characterized by their secretion of IL-9 and IL-10, play a significant role in combating extracellular parasites.244 IL-9 production by Th9 cells is highly dependent on IRF4, which activates the IL-9 gene promoter and regulates IL-9 expression.245,246 IRF4-deficient T cells are hindered in their differentiation into Th9 cells. IRF4 collaborates with various interacting proteins, such as Basic Leucine Zipper ATF-Like Transcription Factor (BATF), EICEs, and SMAD2/3, to regulate Th9 cell differentiation.244
The dysregulation of Th17 cells is closely associated with muptiple diseases, such as psoriasis, inflammatory bowel disease (IBD), and cancers, making them therapeutic targets for regulating immune dysfunctions.247 Th17 and Treg cells both originate from CD4+ naive T cells. Transforming growth factor β (TGF-β) induces naive T cells to differentiate into immunosuppressive Treg cells, while in combination with pro-inflammatory factors like IL-6 and IL-21, it drives the differentiation into immune-promoting Th17 cells.248 An imbalance between Th17 and Treg cells can lead to autoimmune and/or inflammatory diseases. The interplay between IRF4, Th17, and Treg cells is complex. IRF4 deficiency can confer resistance to Th17-related autoimmune diseases due to defects in Th17 cell differentiation.247,249,250 While IRF4 is not essential for Treg production, it is associated with Treg effector functions.244 Treg cells express high levels of IRF4, which can inhibit Th2 cell activity by regulating specific transcriptional programs within Treg cells.251 IRF5 appears to act as a positive regulator in Th17 differentiation. IL-6 stimulates the phosphorylation and nuclear translocation of STAT3 in naive CD4+ T cells, inducing the key transcription factor RAR-related orphan receptor γt expression, which in turn induces IL-17A transcription and drives the Th17-mediated inflammatory response.247,251 IRF5 upregulates IL-6 and STAT3, and inhibits Ikaros and IL-10, promoting the Th17 inflammatory response, while IRF4 negatively regulates this effect of IRF5.236
Follicular helper T cells (Tfh) are a subset of CD4+ Th that are involved in the humoral response, present in secondary lymphoid tissues such as the tonsils, spleen, and lymph nodes.252 Tfh cells are distinguished by their expression of C-X-C chemokine receptor 5 (CXCR5), and programmed cell death protein 1 (PD-1), and they facilitate germinal center B cells differentiation into antibody-secreting plasma cells and memory B cells.253 A deficiency in IRF4 can result in diminished Tfh cell differentiation. IL-21 is a pivotal cytokine in Tfh development, with IRF4 regulating its production and synergizing with the transcription factor STAT3 to control IL-21 signaling.254 IRF4 also cooperates with BATF and BCL6 to regulate Tfh development.255,256
IRFs regulate cell cycle and apoptosis
IRFs are not only integral to the differentiation and development of immune cells but also exert a key role in modulating cell cycle progression and apoptosis. The majority of studies concerning the influence of IRFs on cell cycle and apoptosis have been centered on their role in modulating tumorigenesis. For instance, IRF1 is known to induce the expression of cell cycle inhibitors such as p21 and growth arrest and DNA damage-inducible 45 (GADD45), effectively halting cell cycle progression. In contrast, IRF2 has been shown to facilitate cell cycle processes, acting in opposition to the effects of IRF1. Additionally, IRF3 can directly bind to and activate genes associated with the cell cycle, often in concert with IRF4, thus contributing to the proliferation of myeloma cells.257 IRF5 promotes apoptosis upon signaling via TNF-related apoptosis-inducing ligand (TRAIL) death receptors. This is due to TRAIL-induced IRF5 phosphorylation and nuclear localization, resulting in the transactivation of key death receptor signaling components.258 Multiple studies have demonstrated that IRF5 negatively regulates cell growth and oncogenesis. However, in thyroid malignancies, IRF5 promotes cancer cell proliferation.259 IRF6, as a tumor suppressor gene and transcription factor, functions to suppress tumorigenesis in nasopharyngeal carcinoma (NPC), squamous cell carcinomas (SCCs),and renal clear cell carcinoma.260,261,262 In glioma, IRF6 impaired cell proliferation and induced apoptosis by inhibiting pyruvate kinase isozyme type M2 (PKM2) and glucose transporters 1 (GLUT1) transcription.263 The regulation of tumor development by IRF family members will be extensively reviewed in later chapters.
Furthermore, certain members of the IRF family are crucial to the apoptosis of non-tumor cells. Research has demonstrated that IFN-γ induces apoptosis in primary cultured hepatocytes, and that the presence of IRF1 is necessary for Fas/CD95-mediated cellular apoptosis. Caspase-1/7/8 and TRAIL are potential target genes of IRF1 in the regulation of apoptosis.
Beyond directly impairing virus replication, another critical host defense strategy against viral propagation involves inducing apoptosis in virus-infected cells. IRF3 has dual functions in such cells: it not only activates antiviral genes but also initiates apoptotic cell death through the RIPA pathway.264,265 The activation process of IRF3 in the RIPA pathway is different from its transcriptional activation mechanism, necessitating linear polyubiquitination at particular lysine residues on IRF3 (Fig. 3). In the RIPA signaling pathway, IRF3, upon activation, engages in a critical interaction with the pro-apoptotic protein Bax via the BH3 ___domain proximal to its C-terminal region. The ensuing IRF3-Bax complex undergoes translocation to the mitochondria, acting as a catalyst for the release of cytochrome C into the cytoplasm.266 This event precipitates the subsequent activation of caspases, culminating in apoptosis. However, Otulin, a deubiquitinase that excises linear multiubiquitin chains, suppresses RIPA via deubiquitinating IRF3 in virus-infected cells. To counteract the inhibitory effect of Otulin RIPA facilitates the targeted degradation of Otulin through the continuous action of activated caspase-3 and proteasomes.95 Caspase-3 cleaves Otulin at D31, and the fragment was totally degraded by proteasomes before K48-linked ubiquitination occurs.

Regulation of apoptosis by IRFs. IRF-1 is a tumor suppressor and a regulatory factor of the IFN-γ system, with IFN-γ promoting the expression of IRF-1. IFN-α also induces the rapid phosphorylation and DNA-binding activity of STAT-1, followed by the accumulation of IRF-1 and IRF7. These two transcription factors bind to the TRAIL promoter and induce TRAIL expression. TRAIL is a key participant in the apoptotic pathway and plays a significant role in IFN-induced cell killing. IRF-3 is also involved in the transcriptional induction of TRAIL, where it transactivates the TRAIL promoter upon viral infection, upregulating TRAIL transcription. Conversely, IRF4 actively inhibits the transactivation mediated by IRF1. IRF3 triggers apoptosis through the RIPA pathway, which depends on the linear ubiquitination of specific lysine residues of IRF3. Within the RIPA signaling pathway, IRF3 interacts with the pro-apoptotic protein Bax to form the IRF3-Bax complex and translocates to the mitochondria, catalyzing the release of cytochrome C into the cytoplasm, subsequently activating Caspase, and ultimately leading to apoptosis. Otulin, a deubiquitinase that removes linear ubiquitin chains, inhibits RIPA by deubiquitinating IRF3 in virus-infected cells
Moreover, in a mouse model of alcoholic hepatitis, the non-transcriptional activity of IRF3 regulates the live’s innate immune milieu via increasing immune cells’ apoptotic cell death, thereby promoting the resolution of injury.267
IRF1 and IRF3 regulate PANoptosis
PANoptosis is a novel and unique inflammatory programmed cell death pathway, uniquely regulated by multifaceted PANoptosome complexes.268,269 Previous studies have found that IRF1 is an upstream modulator of PANoptosis, helping to induce the activation of Z-DNA binding protein 1 (ZBP1), NOD-like receptor family, pyrin ___domain–containing 3 (NLRP3), absent in melanoma-2 (AIM2), RIPK1, NLRP12 associated PANoptosis (Fig. 4).268,270,271,272

Regulation of PANoptosis by IRFs. IRF1 has been identified as an upstream regulator of PANoptosis, facilitating the activation of PANoptosome and PANoptosis. To date, molecular characterizations have been conducted on four PANoptosome complexes: ZBP1-PANoptosome, AIM2-PANoptosome, RIPK1-PANoptosome, and NLRP12-PANoptosome. During SARS-CoV-2 infection, innate immune cells produce a variety of inflammatory cytokines, among which the combination of TNF-α and IFN-γ induces PANoptosis. Co-treatment with TNF-α and IFN-γ activates the JAK/STAT1/IRF1 axis, inducing the production of NO and driving caspase-8/FADD-mediated PANoptosis, thereby driving cytokine storms and inflammatory pathology. In the context of hemolytic diseases, heme and PAMPs bind to TLR2 and TLR4, inducing signal transduction through IRF1 and upregulating NLRP12 expression. NLRP12, an innate immune cytosolic sensor, is responsible for inflammasome and PANoptosome activation, inflammatory cell death, and the inflammatory response to heme plus PAMPs, driving inflammatory cell death and pathology under hemolytic conditions. The NLRP12-PANoptosome induces the maturation of IL-1β and IL-18 through caspase-8/RIPK3, driving PANoptosis
IRF1 is involved in the regulation of PANoptosis not only in inflammatory diseases but also in tumors, especially colorectal cancer. During influenza A virus (IAV) infection, ZBP1 protein expression, NLRP3 inflammasome, caspase-1, caspase-8, caspase-3, and mixed lineage kinase ___domain like pseudokinase (MLKL) activation were decreased in IRF1-defective cells. Further studies demonstrated that IRF1 is a transcriptional regulator of ZBP1.271 Genes encoding IRF1, IRF5, and IRF7 are highly upregulated in patients with severe coronavirus disease-2019 (COVID-19), as well as in TNF-α and IFN-γ-treated bone-marrow-derived myeloid cells. Notably, IRF1-deficient cells exhibit resistance to cell death following TNF-α and IFN-γ exposure, a protective effect not observed in cells lacking IRF5 or IRF7.273 Concurrent treatment with TNF-α and IFN-γ triggers the activation of the JAK/STAT1/IRF1 signaling pathway, which in turn stimulates nitric oxide synthesis and promotes caspase-8/FADD-mediated PANoptosis.273 The recent study has uncovered that NLRP12 is a cytoplasmic sensor of heme plus pathogen-associated molecular patterns (PAMPs)-triggered PANoptosis, driving inflammasome and PANoptosome activation, cell death, and inflammation.274 IRF1 is involved in the TLR2/4-mediating signaling pathway to induce Nlrp12 expression, leading to inflammasome assembly and the maturation of pro-inflammatory cytokines IL-1β and IL-18.274 However, IRF1 does not influence inflammation and inflammasome activity but instead acts as an upstream regulator of PANoptosis, inducing cell death during colitis-related tumorigenesis.275 Recently, Zhuang et al. reported that bile acid-induced phosphorylation of IRF3 mediates cell apoptosis through the regulation of ZBP1 gene expression in cholestasis-induced liver and kidney injury.276
IRF6 regulates Keratinocyte development
IRF6 is essential for normal epidermal development and differentiation. Human mutations in the Irf6 gene underlie two genetic conditions: Van der Woude syndrome and popliteal pterygium syndrome.277 Studies have found that mice with null or homozygous missense mutations in the Irf6 gene display a hyperproliferative epidermis incapable of terminal differentiation, leading to abnormal skin, limb, and craniofacial development.278,279 Subsequent research has shown that IRF6 is a key target of Notch in keratinocytes and induces differentiation through a Notch-dependent mechanism.280 Moreover, IRF6 deficiency results in impairing the expression of genes critical for lipids metabolism and formation of tight junctions.73
Signaling pathways IRFs involved
IRFs regulate IFN-I via TLR signaling
TLRs are membrane-bound signal receptors identified in the innate system.281 Structurally, TLRs consist of an extracellular region, a transmembrane region, and an intracellular region.282 So far, 12 functional TLRs have been found in mice and 10 in human.283 TLRs are situated on cell membranes or embedded within endosomes. Engagement with their respective homologous ligands prompts TLRs to recruit adaptors, including MyD88 and/or Toll/IL-1 receptor (TIR) ___domain-containing adaptor protein-including IFN-β (TRIF) to activate IRF proteins alongside other transcription factors (Fig. 5).160

IRFs Respond to TLR Signaling and Participate in the Expression and Function of IFNs. The basal expression of IRF1 maintains the expression of ISGs, thereby conferring antiviral activity. Following viral infection, higher expression of IRF1 is triggered, along with potential cytokines, inducing the expression of IFN and ISGs. Additionally, the secretion of IFN can activate the STAT1-STAT2-IRF9 complex, inducing IRF1 and IRF7 as part of a positive feedback loop. The binding of PAMPs with TLR activates the IRFs signaling pathway, promoting the expression of IFN, pro-inflammatory cytokines, and chemokines, thereby activating the host’s innate immune defense
Specifically, IRF1 is implicated in the TLR7/9-MyD88 signaling cascade, and both IRF3 and IRF7 are involved in the TLR3-MyD88/TLR4-TRIF/MyD88-TBK1 signaling pathways. Additionally, IRF7 is also recruited to the TLR7/9-MyD88 signaling pathway, but IRF3 is not.284,285 IRF5 is involved in TLR7/8/9-MyD88 signaling pathways that lead to the induction of IFN-I genes.
IRF3 and IRF7 can be activated via TLR3/TLR4-TRIF/MyD88-TBK1 signaling pathways. Following ligand recognition, TRIF undergoes phosphorylation and subsequently recruits downstream signaling molecules, including TRAF3, Nucleosome Assembly Protein 1 (NAP1), and TBK1. This cascade facilitates the phosphorylation, dimerization, and nuclear translocation of IRF3 and IRF7 (Fig. 5). Finally induced IFN-I gene expression. Additionally, MyD88/TRAF6-dependent K63-linked ubiquitination of IRF7 is essential for the induction of the IFN-I gene in pDCs by TLR signaling.285
IRF5 is also involved in TLR-dependent (TLR7/8/9) induction of IFNs-I.286,287 The induction of the IFN-I gene via TLR7 and TLR9 is contingent upon the MyD88 adaptor protein. Activation downstream of the TLRs involves TRAF6-mediated K63-linked poly-ubiquitination as well as phosphorylation in the IAD, which is crucial for both IRF5 nuclear translocation and to dislocate a C-terminal autoinhibitory ___domain, thereby facilitating the interaction of activated IRF5 with transcriptional coactivators such as CBP/p300.100,288,289,290,291 Recently studies showed that IRF5 was recruited to endolysosomes by “TLR adaptor interacting with solute carrier family 15 member 4 (SLC15A4) on the lysosome” (TASL) and then phosphorylated by IKKβ or TBK1/IKKε, ultimately inducing IFN-I gene expression(Fig. 5).292,293 This mechanism is an analogy with the IRF3 adaptors stimulator of IFN genes (STING), mitochondrial antiviral signaling protein (MAVS) and TRIF.
IRF1 interaction with MyD88 to control the production of TLR9-dependent IFN-β in mouse DCs.294 IRF1 also be efficiently activated by the TLR-MyD88 pathway, in turn inducing immunity-related GTPase B10 (IRGB10) expression. Moreover, IRF8 engages in the induction of IFN-I genes in TLR-stimulated pDCs but does not interact with MyD88.295 IRF8 in conjunction with TRAF6, modulates TLR signaling, potentially facilitating the interaction between IFN-γ and TLR signal pathways.296 In addition, IRF8 is essential for TLR7- and IFN-α-induced bone marrow differentiation into MDSCs in vitro.297 IRF8 also directly regulates the expression of TLR9 in NK cells. IRF8 regulates hematopoietic stem cells, at least in part, by controlling TLR9 signaling in various innate immune cells.298
IRFs induce IFNs on innate recognition of cytosolic RNA and DNA
RLRs are RNA sensors localized in the cytosol.299 In innate antiviral immunity, except TLR7 and TLR9, the majority of cell types detect viral nucleic acids predominantly via RLRs, thereby initiating antiviral immune responses.300,301 RIG-I and melanoma differentiation-associated gene 5 (MDA5) are two RNA helicase enzymes and essential members of the RLR family. RIG-I and MDA5 both contain two CARD at the N-terminus.300 The MAVS, which contains one CARD ___domain, is the adaptor molecule linking the sensing of viral RNA by RIG-I or MDA5 to downstream signaling.300 MAVS transmits downstream signaling via CARD-CARD interaction.160
IRF3 and IRF7 are critical for the RIG-I/MDA5-mediated IFN-I gene-induction pathway (Fig. 6). Under basal conditions, IRF3 and IRF7 exist in inactive states within the cytoplasm of uninfected cells. After virus infection, TBK1, activated by RIG-I- or MDA5, phosphorylates IRF3 at multiple residues including Ser396, Ser398, Ser402, Ser404, and Ser405 within site 2, which alleviates autoinhibition and allows IRF3 nuclear translocation and interaction with the coactivator CBP.302,303,304 Then, CBP promotes phosphorylation at Ser385 or Ser386 at site 1 within the regulatory region, permitting IRF3 dimerization.303 Presumably, a similar mechanism occurs in IRF7, leading to a holocomplex containing dimerized IRF3 and IRF7, either as a homodimer or heterodimer, and coactivators such as CBP or p300 are formed in the nucleus. These holocomplexes bind to target ISRE DNA sequences within the promoters of IFN-I and IFN-III genes.

PRRs Detect Cytosolic DNA/RNA and Activation of IRFs.Cytosolic RNA or DNA triggers host responses through specific cellular PRRs. The binding of ssRNA or dsRNA to the helicase ___domain of RIG-I/MDA5 induces the activation of RIG-I/MDA5’s CARD and the interaction between the CARD-like ___domain of the adaptor MAVS located on the mitochondrial membrane. This receptor-adaptor interaction leads to the activation of TRAF, TBK1, and IKKε, inducing the phosphorylation of specific serine residues on IRF3 and IRF7. These IRFs then translocate into the nucleus and activate IFN-I genes. NF-κB is also activated and participates in the induction of IFN-I genes. dsDNA is recognized by cGAS, DAI, etc. The adaptor protein STING on the endoplasmic reticulum membrane signals downstream to these DNA receptors, recruiting TBK1 to phosphorylate IRF3, leading to the activation of IFN-I gene expression
Additionally, IRF5 participates in the RIG-I/MAVS signaling pathway.167 Upon infection with vesicular stomatitis virus (VSV) or NDV, IRF5 undergoes translocation from the cytoplasm to the nucleus. However, detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by lung epithelial cells requires MDA5, laboratory of genetics and physiology 2 (LGP2), and NOD1, but not RIG-I (Fig. 6).305 Subsequently, IRF3, IRF5, and nuclear factor kappaB (NF-κB) are phosphorylated and translocate into the nucleus to trigger IFN induction. During SARS-CoV-2 infection, IRF3, IRF5, and NF-κB/p65 are the key transcription factors regulating the IFN response.305
IRF8 is also needed for IFN-I induction in virus-stimulated DCs, which helps to prolong the recruitment of basal transcription machinery to the IFN promoters, a role not shared by IRF7 or IRF3. This supports characteristically high IFN transcription in DCs.295 Moreover, IRF8 and PU.1 collaborate to establish a scaffold complex at the IFN-β promoter, thereby enhancing the recruitment of IRF3 and enabling rapid IFN-β transcription in monocytes.179
Although IRF1 is not critical for the induction of the IFN-I gene by viruses, its expression is usually typically upregulated swiftly after viral infection or poly(I: C) stimulation.306 However, human IRF1 is vital for IFN-γ and STAT1-dependent immunity to mycobacteria.307
Beyond mechanisms for sensing cytoplasmic RNA-, cytoplasmic DNA-sensing systems have also been mentioned (Fig. 6). Known DNA sensors include cyclic CMP-AMP synthase (cGAS), IFN inducible protein 16 (IFI16)/IFN-activated gene box polypeptide 41 (DDX41), IFI204, and (Asp-Glu-Ala-Asp) (DEAD). IRF3 is involved in an antiviral response triggered by double-stranded B-form DNA (B-DNA), and this signaling pathway requires the kinases TBK1 and IKKi.308 ZBP1 is a candidate cytosolic DNA sensor. ZBP1 binds to double-stranded DNA (dsDNA) and, by doing so, enhances its association with IRF3 and TBK1.309 A more recent study indicates that ZBP1 stabilizes Z-form mitochondrial DNA (mtDNA) and facilitates the formation of a cytosolic complex comprising cGAS, RIPK1, and RIPK3 to engage robust IFN-I signaling.310
IRFs involved in the JAK/STAT signaling pathway
The JAK/STAT pathway is a critical cascade in cellular signaling that transmits information from chemical signals outside of a cell to the cell nucleus, resulting in DNA transcription and subsequent gene expression. The interaction between IRFs and the JAK/STAT pathway is not unidirectional. For instance, STAT1 can enhance the expression of IRF1, creating a positive feedback loop that amplifies the immune response.307 Upon viral infection or recognition of PAMPs, IRF3, IRF5, and IRF7 are activated. These IRFs then promote the transcription of IFNs-I (IFN-α and IFN-β) and IFNs-III (Fig. 5). The secreted IFNs engage with their corresponding receptors on cell surfaces, leading to the activation of JAKs. Activated JAKs then phosphorylate the STAT1 and STAT2 leading to the formulation of ISGF3, or IFN-γ-activated factor (GAF). Subsequently, ISGF3 or GAF translocate into the nucleus, where they initiate the transcription of ISGs or GAS respectively and induce IRF1 expression in a positive feedback mechanism, orchestrating a robust antiviral response.307 The IRF9, as part of the ISGF3 complex (together with STAT1 and STAT2), directly binds to the ISREs in the promoters of ISGs, enhancing their transcription.
IFN-II (IFN-γ) binds to cell-surface receptors composed of IFN-γ receptor 1 (IFNGR1) and IFNGR2 subunits, which bind to JAK1 and JAK2, respectively. Activation of these kinases leads to STAT1 homodimerization to formulate GAF, which translocates into nucleus to target GAS DNA sequences and induce IRF1 transcription (Fig. 5).160 In humans, IRF1 is required for the IFN-γ-dependent immunity of macrophage against mycobacteria, whereas its role in IFN-α/β-dependent antiviral immunity is comparatively less critical.307
IRFs involved in the NF-κB signaling pathway
The NF-κB pathway is a key signaling mechanism that controls the transcription of genes involved in immune and inflammatory responses, cell proliferation, and survival. Activation of NF-κB is tightly regulated and can be triggered by various stimuli, including pro-inflammatory cytokines, bacterial or viral infections, and other stress signals. Some IRF members have been shown to interact with the NF-κB pathway, influencing its activity in several ways.
IRF3, IRF5, and IRF7 can function cooperatively with NF-κB to enhance the transcription of certain genes implicated in the immune response. For example, upon viral infection, IRF3, IRF5, and NF-κB can be simultaneously activated and work together to promote the transcription of IFNs-I and other pro-inflammatory cytokines, amplifying the immune response (Fig. 5).305 IRFs can also modulate the activation of NF-κB. For instance, IRF4 has been reported to interact with the NF-κB subunit RelA/p65, affecting its ability to bind DNA and activate transcription. This interaction can either enhance or suppress NF-κB target gene expression, depending on the cellular environment and the specific stimuli.
The role of IRF proteins family in health and diseases
IRFs and infections
Almost all IRFs have been reported to affect pathogenic processes (Fig. 7). Generally, IRF1, IRF3, IRF5, and IRF7 are the main members involved in various microbial infections and microbial-induced human diseases, playing strong roles in IFN-I gene regulation at the early phase of antiviral innate immunity and antibacterial pathogens in most cells.32,63,311,312 Some studies indicate that activation of certain IRFs can also cause excessive inflammatory reactions and body damage,313,314,315 and inhibition of IRFs may offer protection against microbial infections in some certain specific situations.316

The potential role of IRFs in infectious diseases. a The roles of IRFs in viral infections. IRF1 can regulate IFN and inflammasome to fight viral infections, while it also induces cell PANoptosis and virus immune evasion. IRF2, IRF3, IRF4,IRF5, IRF7, and IRF9 play protective roles to prevent viral infections. b The roles of IRFs in bacterial and parasitic infections. IRF1, IRF4, and IRF8 prevent host aginst bacterial infections. IRF3, IRF5, and IRF7 are double-edged swords in bacterial and parasitic infections
Viral infections
Following viral infection, IRFs like IRF1 and IRF3 are dramatically upregulated and activated, inducing endogenous IFN genes such as IFN-α and IFN-β1 production, and/or protein degradation in various cell types, which drive antiviral immunity to multiple clinically important viruses, including hepatitis C virus (HCV), West Nile virus,yellow fever virus, human immunodeficiency virus type 1(HIV1), gamma herpes virus, and human metapneumovirus, and VSV infection.32,165,168,317,318,319,320 IRF1 basal expression provides cellular intrinsic antiviral protection against diverse pathogenic RNA viruses, such as HAV, HCV, dengue virus, and zika virus (ZIKV).321 IRF1 can regulate ZBP1 and NLRP3 inflammasome, mediating inflammatory and cell death responses in IAV infection.271 In addition, IRF1 is required for optimal early activation of IRF3, targeting and augmenting IRF3 phosphorylation, which enhances innate immunity to viral infection.168,322 IRF2 is also identified to inhibit ZIKV and orthopoxvirus replication by enhancing FAM111 trypsin like peptidase A (FAM111A) expression to promote host restriction effect of replication factor C subunit 3 (RFC3).323,324 Moreover, IRF2 is critical for the development and functional maturation of human NK cells to defend against virus-infected cells.325 IRF3 has been reported to trigger apoptosis of virus-infected cells, restricting virus spread within the host, in a RIPA pathway,95,264,326 and IRF3 inhibits NF-κB nuclear translocation to prevent viral inflammation.327 IRF3 and IRF7 are vital for inflammatory responses control during HSV-induced encephalitis (HSE).328 Studies presented that IRF3 and IRF7 deficiency might increase susceptibility to HSE and other respiratory viruses in humans,329,330 and IRF7/9 variants might predispose humans to recurrent, severe HSV or IAV infection.331,332,333 Besides, IRF3 agonist could alleviate Enterovirus A71-induced inflammatory response in mice, indicating a potential therapeutic target in Enterovirus A71-induced hand-foot-and-mouth disease.334 IRF4 is able to promote CD8+ T cell function and exhaustion, limiting memory-like T cells development during lymphocytic choriomeningitis virus (LCMV)-induced infection.335,336 IRF5 is critical for IFN-I and IL-6 induction followed by viral infections,337 and it suppresses HCV replication and induces immune responses in the draining lymph node to defeat West Nile virus infection.312,338 However, in some conditionsmany viruses, including human cytomegalovirus, influenza virus, rhinovirus, EBV, HCV, HIV, and SARS-CoV, can exploit host RNA-binding protein to inhibit expression and functions of IRF1/3/5, thereby preventing a cellular antiviral response.326,339,340,341 Moreover, virus-induced damage and inflammation can activate TLR-IRF pathways, resulting in excessive inflammatory cytokines production and aggravating body damage.313,314
COVID-19, caused by SARS-CoV-2, mainly infects human epithelial cells in the respiratory tract and intestine. Research indicates that patients with SARS-CoV-2 infection have high progesterone levels to facilitate IRF3 phosphorylation, which is associated with decreased severity of COVID-19.342 Nonetheless, SARS-CoV-2 can suppress the IFN-mediated STAT1/2 signaling, diminishing NLRC5, IRF1 and IRF3 gene expression.343,344,345 Furthermore, SARS-CoV-2 papain-like protease and nonstructural proteins (NSP) such as NSP5, NSP12, and NSP13 can cleave IRF3, suppress IRF3 phosphorylation and nuclear translocation to blunt IFN response, which further antagonizes host antiviral innate immunity.346,347,348,349,350,351 On the other hand, a study presented that TNF-α and IFN-γ co-treatment caused the JAK/STAT1/IRF1 pathway activation, leading to caspase-8/FADD-mediated PANoptosis, which resulted in a lethal cytokine shock in mice that mirrored inflammation and tissue damage of COVID-19.273 Shin et al. uncovered that pathological and/or genetic reasons-mediated higher basal IRF1 expression in multiple organs might contribute to the synergistic upregulation of IRF1 under SARS-CoV-2 infection, potentially making individuals more vulnerable to COVID-19.352 Furthermore, Huang et al. illustrated that SARS-CoV-2 activated NF-κB/IRF1 axis to further promote programmed death-ligand 1 (PD-L1) expression and then might facilitate immune evasion, inducing sepsis and fatality.353 Zhang et al. reported that inherent defects in the TLR3- and IRF7-dependent IFN-I immunity may encounter with life-threatening COVID-19 pneumonia in patients without a history of severe infection.354 Besides, Lévy et al. reported an unvaccinated child with life-threatening COVID-19 risk because of an inherited IRF9 deficiency.355 Moreover, it has been found that SARS-CoV-2 spike transfected cells release a large number of microRNAs-loaded exosomes, which are internalized by human microglia and inhibit USP33 expression and IRF9 levels, mediating central nervous system damage through the hyperactivation of human microglia.356
Bacterial and parasitic infections
IRF1 is important for IFN-γ-dependent and TNF-α and IL-6 auto-paracrine signaling loop-mediated macrophagic immunity to mycobacteria.307,357 and IRF1 inhibits mTOR/p70 S6K signaling in macrophages, which facilitates an anti-mycobacterial effect against tuberculosis (TB).358 Furthermore, IRF1 is required for the AIM2 inflammasome activation in response to Francisella novicida (F. novicida) infection,272 and for intestinal group 3 innate lymphoid cells (ILC3s) to produce lots of the protective effector cytokine IL-22 early in the course of Citrobacter rodentium-induced intestinal infection.359 IRF1 also contributes to the innate defense of the cornea against Pseudomonas aeruginosa (P. aeruginosa) infection,360 indicating it is crucial for anatomical containment and prevention of pathogen dissemination. Additionally, the STING/TBK1/IRF3 pathway activation is instrumental in stimulating IFN-I production and inducing apoptosis, thereby offering protection against TB during Mycobacterium bovis (B. tuberculosis) infection.361,362 Furthermore, IRF3, IRF5, and IRF7 are key regulators in the IFN-I-mediated response to mycobacterium tuberculosis (M. tuberculosis).363,364 Specifically, IRF7 facilitates the expression of miRNA-31, which significantly reduces lung pathology and bacilli burdens during M. tuberculosis infection.365 Skjesol et al. found that the Rab11 family interacting protein 2 guided TLR4 sorting adapter translocating chain-associated membrane protein (TRAM) recruitment for orchestrates actin remodeling and IRF3 activation for macrophage phagocytosis of Gram-negative bacteria.366 On the other hand, infections caused by bacteria like Helicobacter pylori (H. pylori) can downregulate IRF3 activation to interrupt STING and RIG-I signaling and then hamper their functions in eliminating bacteria and mobilizing adaptive immune responses.367 A separate study revealed that airway acidification activated the IRF3/IFN-β signaling, subsequently compromising the host’s defensive mechanisms against pulmonary infection by Pseudomonas aeruginosa (P. aeruginosa).315 Besides, mice with IRF3 deficiency were protected from the lethal malaria plasmodium yoelii infection.368 Inflammasome-mediated IRF3 signaling upregulated SOCS1, further inhibiting MyD88-IRF7-mediated IFN-I response in pDCs, inducing parasite fast growth and host death.368,369 Studies have shown that IRF4 expression in T cells is necessary for the effective control of M. tuberculosis and Listeria monocytogenes.370,371 IRF5 has been reported to promote intracellular bacterial clearance and the induction of antimicrobial pathways.372,373 However, IRF5 deficiency ameliorated immunopathology during Salmonella Typhimurium (S. Typhimurium) and Helicobacter hepaticus-induced colitis in vivo.372,374 Moreover, Puthia et al. have illustrated that IRF7 inhibition can be used to prevent and treat bacterial infections other than acute pyelonephritis.316 However, Mathy et al. showed that long non-coding RNA (lncRNA) Nostrill might function as a “guide” to promote recruitment of NF-κB p65 to the IRF7 gene promoter, thereby promoting IRF7 mRNA expression and contributing to intestinal epithelial defense against Cryptosporidium parvum (C. parvum).375 IRF8 activated autophagy genes in macrophages and induced subsequent autophagic capturing and degradation of Listeria antigens to clear Listeria monocytogenes.185 IRF8 also mediated IRF3 phosphorylation and promoted NLRP3 inflammasome activation during infection with Gram-negative bacteria.376
Fungal infections
There are a small number of studies focused on the possible relationships between IRFs and fungal infections. Both A.fumigatus (Aspergillus fumigatus) and C.albicans (Candida albicans) are two major human fungal pathogens. IRF1 induces IRGB10 expression to target the fungal cell wall and cause hyphae damage, modify the A.fumigatus surface and inhibit fungal growth.294 In addition, IRF1 is a fungal recognition pathway downstream and mice lacking IRF1 are hypersusceptible to systemic C.albicans infection.377 Besides, C.albicans infection triggers the cGAS/STING pathway by promoting TBK1 recruitment and activation to phosphorylate IRF3 and mediate IFN-I responses, which further regulate antifungal innate immune responses.378,379 IRF5 is required for Dectin-1-mediated IFN-β production in DCs to defense against C.albicans infection.286 Moreover, IRF5 induces IL-12 production and subsequently generates IFN-γto resist C.albicans, and IRF5 activation blockade significantly increases the susceptibility of Clec2dknockout mice to bloodstream infection with C.albicans.380 Conversely, IRF7 enhances C.albicans infection by regulating CD209 expression and p53-AMPK-mTOR-induced autophagy to inhibit macrophages phagocytosis and killing capacity.381
There are also several researches on the relationships between IRFs and F. monophora (Fonsecaea monophora) infections. For example, F. monophora can triggers Dectin-1 and then activates IRF1 to mediate IL-12 production, which is crucial for antifungal defenses.382 On the other hand, F. monophora has been reported to induce the loss of nuclear IRF1 activity, further blocking IL-12A transcription.382 IRF4 is critical for Treg cell localization and IL-17 expression in Th17 cell to protect against the fungal opportunistic pathogens C.neoformans (Cryptococcus neoformans),383,384 and prostaglandin E2 produced by C.neoformans can inhibit IRF4 function as well as IL-17 production to impede antifungal immunity in T cells.384
IRFs and cancers
Numerous studies have illustrated that the IRF family is important for pathogenesis and therapy of various human cancers (Fig. 8). Traditionally, due to their role in inducing IFN-I to mediate tumors immunosurveillance, several IRFs, such as IRF1, IRF3, and IRF7 are regarded as anti-tumorigenic factors. Conversely, IRF2, which is ascribed as a negative regulator of IRF1, has been recognized as a pro-tumorigenic factor and it drives IFN-dependent CD8+ T cell exhaustion to suppress anti-tumor immunity.8,385 IRF1 and IRF7 knockout mice were shown to have the higher metastasis score than wild-type.386 Besides, activation of the IRF1-X-linked inhibitor of apoptosis-associated factor 1 (XAF1) loop significantly promotes stress-induced apoptosis and restricts the invasive capability of tumor cells.387 However, both pro-tumor and anti-tumor functions have also been illustrated for different IRF members in human cancers.388 It has been reported that IRF1 deficiency in tumor cell presented decreased tumor progression and IRF1 is necessary for PD-L1 upregulation in tumor cells and tumor progression in vivo.389 Conversely, IRF2 is a repressor of PD-L1, and loss of IRF2 in cancers can lead to tumor immune evasion.390 These diverse findings show a complex role for IRFs members in the inflammatory tumor microenvironment.

The potential role of IRFs in diverse common cancers. IRF family is involved in almost all human cancers, with protective roles or negative roles, and some IRF members are double-edged swords in cancers development
Oesophageal cancer
Oesophageal or esophageal cancer primarily comprises esophageal SCC (ESCC) and esophageal adenocarcinoma (EA/EAC).391 Several IRF family members have been reported to be implicated in oesophageal cancers. IRF1 expression level was downregulated while IRF2 was upregulated in ESCC compared with normal esophageal tissues.392 IRF1 suppresses ESCC cell proliferation, while IRF2 acts as an oncoprotein to block the function of IRF1 by preventing its translocation into the nucleus.392 Additionally, IRF2 decreased endogenous IFNGR1 levels and the loss of IFNGR1 turned to increase the resistance of esophageal cancer cells to antitumor IFN-γ.393 Huang et al. revealed that lncRNA IRF1 antisense RNA (lncRNA IRF1-AS) was downregulated in ESCC tissues, and it could repress ESCC proliferation and promote apoptosis. Furthermore, IRF1 directly binds to IRF1-AS promoter to activate IRF1-AS transcription. Therefore, IRF1-AS inhibits ESCC progression by facilitating IFN response via a positive regulatory loop with IRF1.394 Similarly, Watson et al found that IRF1 overexpression inhibited tumor growth in an EA murine model.395 However, a recent investigation described that high levels of forkhead box M1 (FOXM1c) and IRF1 were positively associated with low survival rate and indicated a poor prognosis of oesophageal cancer patients.396 FOXM1c functions as a proliferation-associated transcription factor that is pivotal in driving tumorigenesis and progression of cancer, and is overexpressed in oesophageal cancer patients.396,397 IRF1 is a transcriptional target of FOXM1c and can mediate FOXM1c-related ESCC cell invasion and migration to promote oesophageal cancer metastasis.396 IRF3 single nucleotide polymorphisms (SNPs) may relate to esophageal cancer susceptibility among Chinese patients.398 In addition, the whole-exome sequencing revealed metastatic ESCCs exhibit frequent mutations in multiple genes, including IRF5.399 Additionally, genes like IRF7 may contribute to the susceptibility and/or development of esophageal cancer.400 Sun et al. have identified a positive correlation between the IRF4 protein overexpression and improved outcomes within ESCC.401
In conclusion, IRF1 serves a dual role in esophageal cancer. IRF1 acts as a tumor suppressor to inhibit esophageal cancer cell proliferation. On the other hand, it functions as a transcriptional target of FOXM1c to mediate FOXM1c-related ESCC cell invasion and migration. In addition, overexpression of IRF2 promotes esophageal cancer cell proliferation and mutations of SNPs in genes such as IRF3, IRF5, and IRF7 may facilitate the progress of esophageal cancers, while IRF4 may be regarded as a protective factor in ESCC.
Gastric cancer (GC)
Both IRF1 and IRF2 can restrict GC cell growth and act as protective factors in GC patients. Mutations of the IRF1 gene, inducing the production of functionally impaired IRF1, are critical for GC development.402 In fact, IRF1 can induce apoptosis by upregulation of p53 upregulated modulator of apoptosis (PUMA) in GC cells.403 In addition, IRF1 overexpression can enhance chemosensitivity to 5-fluorouracil in GC cells.404 Recent studies illustrated that IRF1 could suppress DNA damage response (DDR) and reverse chemoresistance by downregulating P-glycoprotein and DNA repair protein RAD51 homolog 1 (RAD51) in GC.405,406 Moreover, IRF1 can inhibit GC metastasis via downregulating microRNA-18a (miR-18a)/miR-19a and Wingless-related integration site (Wnt)/β-catenin signaling.407 Similarly, IRF2 is also regarded as a tumor suppressor in GC, and its downregulation by miR-18a can enhance GC cell proliferation, migration, and invasion.408 Additionally, IRF2 downregulates the expression level of matrix metalloproteinases 1 (MMP1), which promotes GC tumorigenesis, invasion, and metastasis.409 Moreover, overexpressed IRF2 positively affects the prognoses of GC patients and inhibits cancer proliferation by directly promoting the tumor suppressor adenomatous polyposis coli membrane recruitment 1 (AMER1) transcription and inhibiting the Wnt/β-catenin signaling pathway.410 On the other hand, a study showed that IRF3 was a risk factor for GC, and increased IRF3 level was negatively associated with survival in GC patients.411 IRF3 facilitates H. pylori and gastric tumor formation through activating Yes-associated protein (YAP), an important downstream transcription coactivator of Hippo pathway to exacerbate tumor progression.411,412 IRF3 inhibition by amlexanox impedes gastric tumor growth in a YAP-dependent manner.411 Besides, miR-365 inhibited the TLR4/IRF3 axis to reduce IRF3 phosphorylation and YAP expression to restrict gastric precancerous lesions progression.413
DNA methylation of IRF4, IRF5, IRF7, and/or IRF8 frequently occurs in GC cell lines and IRF4 methylation is often observed in GC specimens, which may be a useful biomarker for diagnosing the recurrence of GC.414,415 Furthermore, treatment with demethylating agents restores the expression of IRF4, IRF5, and IRF8, which may result in antitumor activity.414 IRF6 also acts as a tumor suppressor in GC, and its downregulation is clinically correlated with poor prognosis of GC.416 Bioinformatics analysis indicates that IRF6 expression is negatively associated with its promoter methylation in GC cohorts from The Cancer Genome Atlas (TCGA).416 Although Yamashita et al. illustrated IRF5 may exhibit antitumor activity in GC,414 Du et al. indicated that IRF5 was cytoplasm-enriched in GC cells, which promoted GC cell migration by inhibiting expression and inducing degradation of Wnt5a and E-cadherin proteins.417 IRF7 is an indirect target of circular RNA circ0007360 to exert tumor-suppressive role and attenuate GC progression.418 A single-cell RNA sequencing revealed that IRF8 was decreased in CD8+ tumor-infiltrating lymphocytes in GC tissues, and patients with lower IRF8 levels in blood CD8+ T cells likely to be at a more advanced GC stage.419
Overall, IRF1 expression is beneficial for inhibiting the growth of GC cells through inducing tumor cell apoptosis, enhancing sensitivity to chemotherapy drugs, and suppressing the miR-18a/19a and Wnt/β-catenin pathways. In addition, IRF2 can downregulate MMP1 and Wnt/β-catenin pathway, and promote AMER1 transcription to suppress GC proliferation, invasion, and metastasis. Methylation of IRF4, IRF5, IRF6, IRF7, and IRF8 may be associated with GC progression, and demethylation of these factors may increase antitumor activity. However, cytoplasmic IRF5 also can inhibit Wnt/E-catenin and then facilitate GC migration. Furthermore, IRF3 acts as a tumor promoter by activating YAP to exacerbate GC progression.
Liver cancer
Primary liver cancer can mainly be subdivided into hepatocellular carcinoma (HCC), intrahepatic cholangiocarcinoma, and other rare types.391 IRF1 in HCC pathogenesis is bidirectional, serving as both a tumor promoter and suppressor. On the one hand, IRF1 upregulates PD-L1 expression, which is beneficial for HCC cells to escape from anti-tumor immunity.420 IRF1 can activate mTOR/STAT3/ AKT serine/threonine kinase (AKT) signaling, upregulating Slug, Snail, and Twist to promote HCC cell invasion, migration, and epithelial-mesenchymal transition (EMT).421 IRF1 also can activate human endogenous retrovirus-H long terminal repeat-associating 2 (HHLA2) by binding to the promoter region of HHLA2, which induces macrophages M2 polarization and chemotactic migration, leading to HCC immune escape and development.422 Studies on the suppression of IRF1 activity may target HCC treatments. For example, the activity of IRF1 can be reversed by IL-33 SUMOylation, resulting in impaired antitumor immunity in HCC cells.423 The overexpression of miR-130b and miR-345, which induces the suppression of IRF1, can markedly inhibit HCC cell migration and invasion.421,424 However, IRF1 also can promote autophagy, cell death, and inhibit growth in HCC cells and mice.425,426 It has been reported that the repression of IRF1 may facilitate the progression from chronic HCV to cirrhosis and HCC.427 IRF1 has the tumor-suppressor effect and higher expression of IRF1 is associated with better outcomes in HCC patients.428,429 In addition, Yan et al. revealed that IRF1 induces miR-195 to impede checkpoint kinase 1 (CHK1) protein expression, and subsequently upregulates HCC cellular apoptosis, while IRF1 expression or CHK1 inhibition facilitates PD-L1 expression by increased STAT3 phosphorylation.430 Furthermore, a recent study discovered that IRF1 positively regulates C-X-C motif chemokine ligand 10 (CXCL10) and CXCR3 to induce CD8+ T cells, NK, and NK T cells enrichment and migration, and IFN-γ secretion, causing HCC cells apoptosis, thus overcoming proliferative effects from IRF1-induced PD-L1 expression.431 Besides, Studies reported that miR-31, miR-23a, and miR-301a may induce HCC progression via the downregulation of IRF1.432,433,434 Nuclear receptor subfamily 4 group A member 1 (NR4A1) mediates NK cells dysfunction in HCC by inhibiting the IFN-γ/p-STAT1/IRF1 signaling, and NR4A1 deficiency restores the cytotoxicity of NK cells and improves anti-PD-1 therapy efficacy.435
Although Guichard et al. once identified that IRF2 might be a tumor suppressor gene in HCC and IRF2 inactivation impaired TP53 function,436 other studies revealed that high expressed of IRF2 was relevant to increased recurrence probability in HCC patients and IRF2 downregulation resulted in significantly decreased invasion ability, combined with decreased STAT3, p-STAT3, and MMP9 in HCC cell lines.428 IRF2 was found to promote proliferation and inhibit apoptosis of HCC, and increase lenvatinib resistance by upregulating β-catenin, indicating that inhibiting IRF2 is a potential strategy to enhance the therapeutic effect of lenvatinib on HCC.437 Moreover, major vault protein induces human double minute 2 to liberate from IRF2, which causes P53 ubiquitination and then promotes HCC development.438 In addition, as a competitive antagonist for IRF1, overexpressed IRF2 can downregulate CXCL10 expression via the inhibition of IRF1/CXCL10/CXCR3 axis and decrease PD-L1 expression via the inhibition of IFN-γ/IRF1 to partially inhibit anti-tumor activity of IRF1 in HCC.420,431
The role of IRF3 in HCC has also been reported with varying outcomes. Yuan et al. showed that the survival rates in HCC patients improved significantly as a function of increased TLR3 and IRF3 expression levels.439 Moreover, IRF3 was positively associated with TLR3 expression in HCC tissues, which may present a synergistic effect on apoptosis and restrain HCC cells proliferation, MMP2 expression, and angiogenesis.439 On the other hand, other studies have illustrated that IRF3 has the potential to promote HCC progression. Overexpressed IRF3 was markedly correlated with clinical stages and pathological grades in HCC.440 HBV-mediated N6 methyladenosine epitranscriptomic regulation of phosphatase and tensin homolog (PTEN), a well-known tumor suppressor, affects innate immunity by inhibiting IRF3 nuclear import and facilitates HCC development by activating the phosphatidylinositol 3-kinase (PI3K)/AKT pathway.441 Ionizing radiation, the main reason for the failure of radiotherapy, can activate the TBK1/IRF3 pathway, upregulating PD-L1 in HCC cells and restricting CTLs activity and protecting HCC cells from immune-mediated eradication.442 Disruption of the BRCA1 DNA repair associated-partner and localizer of BRCA2 (PALB2) interaction causes persistent DNA damage in HCC cells, activating the cGAS/STING signaling, which induces PD-L1 expression via the STING/IRF3/STAT1 pathway and then suppresses immune to facilitate HCC tumorigenesis and progression.443
Yuan et al. revealed that overexpressed IRF4 restricted HCC cell proliferation and migration capacity by inhibiting the JAK2/STAT3 signaling pathway and EMT.444 Methylation arrays revealed that IRF5 and IRF7 were frequently hypermethylated in HCC tissues.445,446 Furthermore, IRF5 was downregulated in livers of HCV-positive compared to HCV-negative HCC patients or healthy controls, and IRF5 suppressed HCV replication and HCC pathogenesis.312 The expression of IRF8 was also decreased in HCC samples, and IRF8 overexpression in HCC cells significantly improved antitumor effects in immune-competent mice via regulating the recruitment of tumor-associated macrophages (TAMs), inhibiting CCL20 expression, and promoting anti-PD-1 therapy response.447 Besides, it has been reported that IRF9 overexpression promoted IFN-induced activity of dsRNA-dependent protein kinase (PKR) in human hepatoma cells.448 Reduction in IRF9 levels caused resistance to IFN-α2a treatment in HCC cells,449 and another study showed that disease-free survival (DFS) and overall survival (OS) were better in IRF9-positive versus IRF9-negative patients with HBV-related HCC who received postoperative IFN-α treatment, indicating that IRF9 can act as a predictive marker of outcome after postoperative IFN-α therapy in HBV-related HCC patients.450
In a word, the roles of IRF1, IRF2, and IRF3 in HCC are complex and bidirectional, exhibiting both tumor-promoting and anti-tumor effects. IRF1 upregulates PD-L1, and HHLA2, and activates the mTOR/STAT3/AKT pathway to facilitate HCC immune escape, invasion, and migration; meanwhile, IRF1 also enhances HCC autophagy and induces miR-195, CXCL10, and CXCR expression to cause HCC cell apoptosis and death. Although IRF2 positively regulates TP53 function to inhibit HCC growth, it also induces β-catenin expression and inhibits IFN-γ/IRF1 and IRF1/CXCL10/CXCR3 axis to suppress HCC apoptosis, enhance HCC proliferation and resistance to therapeutic drugs (Lenvatinib). IRF3 contributes to HCC apoptosis and inhibits HCC proliferation, yet it also causes HCC progression via upregulating PD-L1 expression. IRF4, IRF5, IRF8, and IRF9 act as protective factors to suppress HCC progress.
Pancreatic cancer (PC)
PC is mainly classified into exocrine and neuroendocrine neoplasms, of which pancreatic ductal adenocarcinoma (PDAC) accounts for >85% of pancreatic tumors.451 IRF1 and IRF2 can regulate PC progression by functioning as an anti-oncoprotein and oncoprotein, respectively.452 Additionally, IRF1 facilitates 2'3’-cGAMP/BV6/zVAD.fmk-induced necroptosis in PC.453 Furthermore, the zinc finger protein ZBED2, essential for PDAC progression, can antagonize IRF1-induced transcriptional activation and suppress the pancreatic progenitor transcriptional program, promotemotility, and enhance invasion in PDAC cells.454 These studies illustrate that IRF1 is a tumor suppressor in PC. Instead, IRF2 promotes PC tumorigenesis, and Cui et al. revealed that IRF2 was upregulated in primary PC tissues and was associated with tumor differentiation, size, tumor-node-metastasis stage, and survival of PC patients.455 IRF2 inhibits apoptosis effectors to facilitate PC cell growth.455 PC patients with high IRF2 and IRF6, and low IRF3 levels in tumors had significantly poorer OS.456 Similarly, a study based on bioinformatics database and proteomics reported that PDAC patients with lower IRF3 expression had shorter OS.457 However, another study described that IRF3 promoted circRNA ubiquitin-like with PHD and RING finger ___domain 1 (circUHRF1) expression, which can regulate ADP ribosylation factor like GTPase 4 C (ARL4C) expression and then induce PDAC progression by sponging miR-1306-5p.458 The role of IRF6 in PC is currently unclear, Xie et al. revealed that IRF6 might be one of the risk genes in pyroptosis-related genes (PRG), and high PRG prognostic index had a lower probability of survival in PC patients based on the TCGA dataset.459
A recent study showed that IRF4 played a critical role in shaping the immune cell composition within the tumor microenvironment (TME) of murine PC and deficiency of IRF4 accelerated tumor growth and reduced survival, combined with a dense tumor infiltration with polymorphonuclear myeloid-MDSC (PMN-MDSC) and reduced numbers of CD8+ T cells.460 Similarly, the repression of IRF5 and IRF7 promotes MYC proto-oncogene, bHLH transcription factor (MYC)/ KRAS proto-oncogene, GTPase (KRAS)-dependent evasion of B cells and NK cells in PDAC, and the de-repression of IRF5 and IRF7 results in increased survival.461 Furthermore, Meyer et al. revealed that reduced IRF8 level in circulating pre-DCs was correlated with decreased OS and relapse-free survival (RFS) in PC patients.462
In conclusion, IRF1 can suppress mitochondrial respiration and fatty acid synthesis in PC cells and induce PC cell necroptosis. In addition, the inhibition of IRF4, IRF5, IRF7, and IRF8 decreases the survival of PC patients, indicating these IRFs probably act as anti-tumor factors in PC. On the other hand, IRF2 and IRF6 may function as oncogenic proteins due to they are negatively related to PC patients’ survival, and IRF2 can inhibit PC cell apoptosis. Although lower IRF3 is associated with shorter OS for PDAC patients, it upregulates circUHRF1 to induce PDAC progression.
Colorectal cancer (CRC)
IRF1 is a protective factor and functions as a tumor suppressor to prevent CRC progress. Hong et al. revealed that IRF1 expression was reduced in CRC and IRF1 level was inversely associated with CRC metastasis.463 A study based on bioinformatics methods indicates that CRC patients with higher IRF1 expression presented better RFS time, and the DNA methylation in the IRF1 gene region is indicated to be negatively correlated with IRF1 expression and positively correlated with CRC recurrence.464 Moreover, IRF1 can promote Ras association ___domain-containing protein 5 (RASSF5) expression, and then suppress CRC proliferation and metastasis, possibly through downregulating the RAS-Rac family small GTPase 1(RAC1) pathway.463 IRF1 positively regulates PANoptosis to restrict tumorigenesis in CRC.275 More recently, Yuan et al. found that IRF1 reduces autophagy related 13 (ATG13) expression to inhibit autophagy and induce apoptosis, which further prevents CRC progression.465 Besides, upregulated IRF1 can promote the radiosensitivity of CRC both in vitro and in vivo.91
Sun et al. reported that CRC patients with high exosomal IRF2 content had a poorer OS rate compared to patients with lower exosomal IRF2 content.466 Exosomal IRF2 can induce vascular endothelial growth factor C (VEGFC) release by macrophages, and inhibition of IRF2 attenuates the lymphatic network remodeling in the sentinel lymph node (SLN) and suppresses the SLN metastasis in CRC.466 However, Liao et al. revealed that CRC patients with low IRF2 expression may be non-responders to anti-PD-1, while patients with high IRF2 expressional most presented a complete response, partial response, or stable disease.467 Furthermore, IRF2 suppresses the migration and infiltration of MDSCs by repressing the CXCL3/CXCR2 axis, and IRF2 overexpression in microsatellite instability-high (MSI-H) CRC overcomes the resistance of tumors, expressing KRAS to anti-PD-1 therapy.467
IRF3 acts as both a tumor promoter and suppressor in different studies. A bioinformatic investigation reported that IRF3 was upregulated in CRC and suggested a shorter survival time in CRC patients.468 Besides, another study revealed that drugs like wogonin exhibit anti-tumor activity against CRC by downregulating IRF3 expression.469 However, Tian et al. identified IRF3 as a tumor suppressor and a prognosis marker for CRC patients.470 IRF3-deficient mice are more susceptible to the development of intestinal tumors and IRF3 negatively regulates the Wnt/β-catenin pathway to prevent tumorigenesis in CRC.470 In fact, β-catenin may exert oncogene activity in part through impeding the nuclear translocation of IRF3 and then inhibiting the IRF3-mediated immune signaling pathway in CRC cells.471,472 Similar to IRF3, the role of IRF7 in CRC development is also controversial. Chen et al. reported that IRF7 was also upregulated in CRC and suggested a shorter survival time in CRC patients.468 On the other hand, evidence showed that tumor-infiltrating pDCs with positive IRF7 expression were associated with prolonged survival of CRC patients,473 and 5-aza-2-deoxycytidine exhibited anti-tumor efficacy via activating MDA5/MAVS/IRF7 pathway in CRC-initiating cells,474 suggesting that IRF7 may have an anti-tumor effect in CRC.
There are a few researches focused on the role of IRF4, IRF5, IRF6, IRF8, and IRF9 in CRC. Wang et al. revealed that IRF4 was downregulated in colon cancer tissues and that overexpression of RNF2 could increase the ability of proliferation, migration, and invasion in colon cancer cells (SW480 cells) by promoting the ubiquitination and degradation of IRF4.96 Moreover, Udden et al. showed that NOD2 plays a vital role in the suppression of inflammation and tumorigenesis in the colon via inducing IRF4 and then downregulating the TLR signaling pathways.475 Integrated multi-omics analyses identified that patients who carried concurrent IRF5 and NEFH mutations had worse survival outcomes.476 Hu et al. presented that IRF5 probably was a significant mediator of DNA damage signaling and the induction/activation of IRF5 signaling pathway might enhance CRC cell death with chemotherapeutic agents.477 In addition, Arnold et al. revealed that GM-CSF induced IRF5 activation in eosinophils and then promoted antitumor immunity in CRC.478 Tan et al. found that IRF6 expression was reduced in CRC and liver metastasis, and IRF6 enhanced CRC cell sensitivity to cisplatin to inhibit cell proliferation, migration, and invasion along with aggravating cell apoptosis.479 Ibrahim et al. described that human colorectal carcinomas had significantly lower IRF8 expression and higher IRF8 promoter DNA methylation compared to normal colon, and mice with Irf8 deleted in colonic epithelial cells showed higher inflammation-induced tumor incidence.480 IRF8 can repress osteopontin expression in colon epithelial cells, and IRF8 expression is silenced during the transformation of colon epithelial cells into colon tumor cells, indicating that tumor cells may inhibit IRF8 to upregulate osteopontin, which functions as an immune checkpoint to restrict CTL activation and promote host tumor immune tolerance and evasion.481 On the other hand, IRF9 may promote CRC development by activating IL-6/STAT3 pathway in CRC models.482
These evidences illustrate that IRF1, IRF4, IRF5, IRF6, and IRF8 may be candidate tumor-suppressors in CRC. IRF1 enhances RASSF5 expression and CRC cells PANoptosis as well as radiosensitivity to suppress CRC tumorigenesis and progression. IRF5 induces CRC cell DNA damage, and cell death, and enhances antitumor immunity. IRF6 contributes to CRC cell apoptosis and enhances sensitivity to cisplatin chemotherapy. However, the roles of IRF2, IRF3, and IRF7 in CRC are controversial. Although exosomal IRF2 induces VEGFC release and the inhibition of IRF2 suppresses SLN metastasis in CRC, IRF2 also inhibits the CXCL3/CXCR2 axis to prevent CRC, and its overexpression reverses CRC cells’ resistance to anti-PD-1 treatment. Similarly, high expression of IRF3 and IRF7 led to poor survival time in CRC patients. On the other hand, IRF3 suppresses Wnt/β-catenin to inhibit CRC tumorigenesis and IRF7 activation induces anti-tumor activity in CRC cells.
Lung cancer
Lung cancer is divided into two broad categories: the common non-small cell lung cancer (NSCLC) and the relatively rare SCLC.483 NSCLC can be further divided into lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and large cell carcinoma (LCC).484 Decreased IRF1 has been reported in NSCLC tissues,485,486 and inhibition of the IRF1/IFN-γ pathway by phosphoserine aminotransferase 1 promotes LUAD metastasis.487 Frequent genomic aberrations of IRF1 were also found in LUAD metastatic samples by regulator network analysis based on TCGA.488 IRF1 acts as a prognostic factor for NSCLC and acts as a potential tumor suppressor by suppressing oncogenic protein Karyopherin alpha 2 (KPNA2) gene expression in LUAD cells.485 Zhang et al. revealed that the first-line NSCLC chemotherapy drug cisplatin, could induce IRF1 activation in vitro, which further enhanced mitochondrial depolarization, oxidative stress, apoptotic cell death, and suppressed autophagy in NSCLC A549 cells,486 indicating that IRF1 is a possible target for promoting the sensitivity to cisplatin therapy in NSCLC.
The possible function of IRF2 in lung cancer is unclear. A few studies demonstrated that both miR-18a-5p and miR-1290 promote NSCLC proliferation and invasion by suppressing IRF2.489,490 Moreover, lncRNA GAS5 and CASC2 can inhibit the progression of NSCLC and repress cisplatin resistance of NSCLC in vitro and in vivo through inhibiting miR-221-3p and miR-18a, thus increasing IRF2 expression in NSCLC cells, respectively.491,492 However, another study reported that miR-450 could inhibit lung cancer progress by downregulating IRF2 in vitro and in vivo.493 These researches indirectly indicate that IRF2 acts as both a protective and risk factor in NSCLC.
Yi et al. revealed that overexpressed IRF3 induced the apoptosis of NSCLC cells and phosphorylated IRF3 (p-IRF3) was decreased in NSCLC samples.494 Gong et al. presented that inhibiting mutant epidermal growth factor receptor (EGFR) activated the RIG‑I/TBK1/IRF3 pathway and then resulted in a tumor-suppressive role in NSCLC with EGFR-activating mutations.495 Zhang et al. found that Niraparib and radiation synergistically inhibited tumor immunity in EGFR-mutated NSCLC both in vitro and in vivo, accompanied by increased CD8+ T lymphocytes and the STING/TBK1/IRF3 pathway activation.496 Similarly, other studies described that inhibition of WEE1 G2 checkpoint kinase (WEE1) or use of carboplatin or other DDR inhibitors could activate the STING/TBK1/IRF3 pathway, promoting the antitumor immune response to PD-L1 blockade in lung cancer models.497,498,499 In addition, Long et al. illustrated that G protein-coupled receptor 162 could act as a novel radiotherapy sensitizer by interacting with STING, which then promoted DDR and activated IRF3 to inhibit the occurrence and development of tumors in lung cell lines.500
Some studies reported that IRF4 in peripheral mononuclear cells was a protective prognostic factor in NSCLC patients and LUAD patients with high IRF4 expression correlated with significantly longer OS.501,502 Another study pointed out that IRF4 in tumor tissues was associated with poorer survival of NSCLC patients.503 In fact, IRF4 facilitates macrophages M2 polarization, and the E74 like ETS transcription factor 4 (ELF4)/IRF4 pathway activation by exosomal circZNF451 in LUAD patients reshapes the immunosuppressive TME and impedes the effect of PD-1 blockade treatment.504 IRF4 downregulation by liver X receptors (LXR) contributes to the anti-tumoral effects of LXR activation in lung carcinoma models.505 Besides, thigh-dimensional single-cell profiling of T cells from chemotherapy-naive patients with NSCLC revealed that IRF4+ CD4+ effector Tregs suppress antitumor immunity through expressing molecules related to enhanced immunosuppression.506
Feng et al. found reduced IRF5 expression in human NSCLC tissues,507 while Guo et al. revealed that IRF5 level was significantly higher in the peripheral blood of NSCLC patients.508 Yamashina et al. identified IRF5 as a specific factor for cancer stem-like cells from chemoresistant tumors and it was important for M-CSF and tumorigenic myeloid cellsgeneration. IRF5/M-CSF pathway activation in tumor cells was correlated with the number of tumor-associated CSF1 receptor+ M2 macrophages in NSCLC patients.509 IRF6 level was also reported to be upregulated in both LUAD and LUSC tissues, and inhibition of IRF6 revealed the antitumor effects in lung cancer cells.510 Huang et al. demonstrated that higher IRF7 levels in LUAD tumors were associated with higher microvessel density in LUAD tissues, potentially responsible for the hemorrhage outcomes following bevacizumab treatment.511 Besides, IRF7 can enhance constitutive PD-L1 expression though directly promoting transcription of PD-L1 and tumor cell immune evasion, and methylated IRF7 is negatively correlated to PD-L1 expression in NSCLC.512
Suzuki et al. revealed that the IRF8 methylation level was significantly higher in NSCLC specimens compared to non-malignant lung tissues, and that IRF8 methylation may act as a prognostic marker for NSCLC recurrence.513 In addition, Liang et al. showed that IRF8 expression was frequently diminished in lung tumoral tissues, and IRF8 induced lung cancer cell senescence through inhibiting AKT signaling and promoting P27 protein accumulation.514 However, survival analyses based on the TCGA database reported that LUAD patients with higher expression levels of IRF8 were associated with poor survival.515 IRF9 has been reported to be overexpressed in lung cancer and is associated with poor survival, promoting lung cancer progression via the upregulated expression of oncogene versican.516
In conclusion, IRF1 and IRF3 present inhibitory functions in lung cancer. IRF1 activation induces lung cancer cell death and suppresses KPNA2 expression to inhibit LUAD growth. IRF1/IFN-γ inhibition facilitates LUAD metastasis. IRF3 contributes to NSCLC apoptosis and the activation of RIG-I/STING/TBK1/IRF3 pathway inhibits NSCLC growth and increases antitumor immunity. Conversely, IRF5, IRF6, IRF7, and IRF9 may induce lung cancer progression. IRF5/CSF activation facilitates tumor-associated M2 expression, and IRF7 induces PD-L1 production. IRF9 increases versican expression,which is associated with poor survival in lung cancer patients. Decreased IRF6 showed an antitumor effect. The detailed functions of IRF2, IRF4, and IRF8 in lung cancer are complex and controversial. On the one hand, IRF2 inhibition by miR-18a-5p and miR-129 induces NSCLC proliferation and invasion. However, IRF2 inhibition by miR-450 suppresses lung cancer cells’ progress. IRF4 overexpression causes poor survival in NSCLC patients, and ELF4/IRF4 axis activation causes M2 polarization to promote LUAD. Inhibition of IRF4 facilitates the LXR anti-tumor effect; while other studies have shown that IRF4 expression contributes to long OS in lung cancer patients. IRF8 has been reported to suppress ATK and induce P27 expression to cause lung cancer cell senescence, and IRF8 methylation leads to NSCLC recurrence. Another report showed that IRF8 upregulation was associated with poor survival of LUAD patients.
Ovarian cancer (OC)
Transcriptome analysis identified IRF1 was associated with platinum sensitivity, and its overexpression was correlated with increased OS and progression-free survival in high-grade serous OC (HGSOC).517 However, another study revealed that IFN-γ induced IRF1 expression, which was followed by increased PD-L1 expression in OC cells, and IRF1 inhibition attenuated the IFN-γ-induced gene and PD-L1 protein levels. Moreover, IFN-γ-induced PD-L1 expression is modulated by the JAK1/STAT1/IRF1 pathway in OC.518 Additionally, activation of the TLR4/IL-6/IRF1 signaling causes androgen receptor (AR) upregulation and taxol resistance genes transactivation in OC.519 Furthermore, the chemotherapeutic drug cisplatin upregulates IRF1 expression, which in turn limits the cell response to cisplatin in OC cells,520 indicating that IRF1 is a potential target in OC chemoresistance and progression.
Zhang et al. showed that deubiquitinase USP35 was significantly elevated in cisplatin-resistant OC cells, partially inhibiting the STING/TBK1/IRF3 pathway and then resisting the antitumor effect of IFN-I.521 On the other hand, POL I inhibitor CX-5461 activates cGAS/STING/TBK1/IRF3 pathway, increasing immunotherapy efficacy in OC.522 The immunostaining analysis showed that IRF4 was expressed in most HGSOC patients;523 however, the possible function of IRF4 on OC remains unclear. Zhang et al. illustrated IRF5 overexpression in combination with its activating kinase IKKβ reprogramed TAMs to a phenotype that induced anti-tumor immunity and promote tumor regression in OC model.524 In addition, IRF9 was reported to be the critical upstream regulator to mediate growth-inhibitory effects of IFN-α on OC cells, and the anti-tumoral effect of chemerin on OC cells in vitro was regulated by the activation of IFN-α response genes via IRF9.525,526 Therefore, IRF3, IRF5, and IRF9 may be protective and function as anti-tumor factors in OC.
Prostate cancer (PCa)
Adenocarcinoma is the major type of PCa.527 Bioinformatics analysis revealed that IRF1 was a tumor suppressor within this core transcriptional regulatory network in modulating PCa cell proliferation.528 IRF1 was also associated with necroptosis in the prostate adenocarcinoma cell line PC-3 cells,529 and oncogenic factor the Deltex-3-like E3 ubiquitin ligase (DTX3L) can induce proliferation and survival of metastatic PCa cells via the inhibition of IRF1.530 Wu et al. identified that IRF8 expression was increased in primary PCa but reduced in castration-resistant prostate cancer compared with normal prostate tissue.531 IRF8 promoted the degradation of the AR and decreased IRF8 facilitated AR-induced PCa progression and enzalutamide resistance, indicating that IRF8 may act as a tumor suppressor in PCa pathogenesis and represent a potential therapeutic option to defeat enzalutamide resistance.531
Although the detailed function of IRF2, IRF3, and IRF7 in PCa remain unclear, studies demonstrated that miR-221 could inhibit cell growth and invasiveness, and induce cell apoptosis, in part by suppressing IRF2 and SOCS3 in PCa cells, and anti-cancer drug nobiletin inhibits PCa cell growth through suppressing TLR4/TRIF/IRF3 and TLR9/IRF7 pathways.532,533 On the contrary, Zhao et al. illustrated that IRF7 overexpression in PCa cells had a marked effect on inhibiting bone metastases in xenograft nude mice.534
Renal cancer
IL-4 was reported to increase IRF1 expression, which then inhibited the proliferation of human renal cell carcinoma (RCC) cell lines.535 Tomita et al. demonstrated that IFN-γ induced STAT1/IRF1 activation, followed by caspase-7-mediated apoptosis in RCCs cell line ACHN cells.536 However, with IFN-γ stimulation, activation of JAK2/STAT1/IRF1 signaling also induced PD-L1 expression in both WT- and Mut-VHL clear cell RCC (ccRCC) cells.537 IRF3 was overexpressed in ccRCC and significantly associated with worse clinical outcomes and adverse OS in ccRCC patients based on databases.538 The multi-omics analysis on ccRCC samples extracted from Gene set enrichment (GSE) and TCGA data sets identified IRF4 as a protective factor in ccRCC.539 Moreover, Wang et al. illustrated that Zinc finger protein 692 induced tumorigenesis in ccRCC via the transcriptional repression of IRF4 and other genes.540 IRF6 expression was reduced in ccRCC tissues and cell lines, and downregulated IRF6 was associated with worse clinicopathological features and poorer prognosis.541 IRF6 inhibited ccRCC cell proliferation, invasion, and migration at both the cellular and animal levels which may be through the inhibition of kinesin family member 20 A (KIF20A).262 Studies illustrated that IRF7 was one of upregulated differentially expressed genes in ccRCC samples and patients with high IRF7 expression presented a relatively worse survival rate.542,543 CircEGLN3 sponged miR-1299 to enhance the IRF7 level, which is responsible for RCC cell proliferation and aggressiveness in vitro.543 It was reported that ccRCC patients with high IRF8 expression within metastatic sites had longer OS than those with low IRF8 expression.544 IRF8 was identified to inhibit RCC cell colony formation and migration through promoting cell cycle G2/M arrest and apoptosis, upregulating tumor suppressor genes like caspase-1, p21, and PTEN expression, and inhibiting oncogenes YAP1 and survivin expression.545 However, Nixon et al. revealed that TAMs expressed IRF8 to promote T cell exhaustion in RCC and other cancers, and ccRCC patients with abundant CD8+ T cell infiltration might have worse survival if they expressed higher IRF8-TAM gene signature.546
In conclusion, IRF1 and IRF8 have dual effects on renal cancers. IRF1 can inhibit RCC proliferation, and STAT1/IRF1 activates caspase-7-mediated apoptosis in RCC cells; meanwhile, the JAK2/ STAT1/IRF1 pathway also induces PD-L1 expression in ccRCC cells. Similarly, IRF8 induces G2/M arrest and cell apoptosis, upregulates tumor suppressors p21, and PTEN and inhibits oncogenic YAP1 and surviving, thus suppressing RCC cell colony formation and migration; but IRF8+TAMs promote T cell exhaustion, which is associated with worse survival in ccRCC patients. IRF3 and IRF7 may be oncogenic factors due to they are associated with worse outcomes and survival in ccRCC patients, and IRF7 facilitates RCC cell proliferation and aggressiveness. On the other hand, IRF4 and IRF6 are tumor suppressors. ZNF692 represses IRF4 to induce ccRCC tumorigenesis and IRF6 inhibits cRCC cell proliferation, invasion, and migration.
Leukemia
The most common leukemias are acute myeloid leukemia (AML), acute lymphocytic leukemia/lymphoma (ALL), chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), and CML. There are also some relatively rare leukemias, such as chronic myelomonocytic leukemia (CMML) and adult T-cell leukemia/lymphoma (ATL). IRF1 plays a vital role in myeloid differentiation by acting as a tumor suppressor gene in leukemia, and mutations in IRF1 are likely to influence IRF1 and its DNA binding affinity, leading to the inhibition of cancer suppression.547 Semmes et al. identified IRF1 SNP as one risk locus in ALL.548. Massimino et al. showed that IRF5 exerted antiproliferative effects, inhibited B-cell receptor (BCR)-ABL signaling, and increased the cytotoxicity of imatinib mesylate in both immortalized and primary CML cells.549 Mathew et al. presented that with sorafenib treatment, active IRF7 (p-IRF7/t-IRF7) levels were increased in both mouse and human leukemia cells, which then facilitated IL-15 production. This, in turn, promoted immune memory against tumor cells, inducing an immune-mediated cure of AMLFLT3-ITD-relapse.550 Moreover, IRF7 overexpression decreased AML cell proliferation and leukemia stem cell levels, while IRF7 knockout accelerated AML progression and induced vascular cell adhesion molecule 1 (VCAM1)- very late antigen 4 (VLA-4) mediated intracerebral invasion in AML mouse models.551 In addition, IRF7/stress-activated protein kinase (SAPK)/JNK pathway activation resulted in more M1 characteristics and contributed to prolonged survival in leukemia mice.552 Tian et al. revealed that both IRF9 mRNA and protein levels were decreased in human AML samples.553 IRF9 is a negative regulator for AML, which binds with the SIRT1 promoter, represses SIRT1 expression, and promotes p53 expression to inhibit AML cells proliferation, colony formation, and survival.553 These evidences suggest that IRF1, IRF5, IRF7, and IRF9 are tumor suppressors in leukemia.
However, studies revealed that IRF2 and IRF3 act as oncoproteins in leukemia. IRF2 promotes inositol polyphosphate-4-phosphatase, type-II (INPP4B) expression, which then facilitates AML cell growth and colony formation, and IRF2/INPP4B pathway activation represses apoptosis through induction of autophagy combined with the inhibition of Th1 cell differentiation and promotion of Th2 cell differentiation in AML cells.554,555,556 In addition, the inhibition of IRF2 and the IRF2/INPP4B axis by miR-222-3p exposed to human bone marrow mesenchymal stem cells (hBM-MSCs-Exo) suppressed proliferation and promoted apoptosis of AML cells; while overexpressed either IRF2 or INPP4B resisted the proliferation-inhibitory and pro-apoptotic effects mediated by BM-MSCs-Exo.557 Tian et al. showed that both IRF3 mRNA and protein levels were significantly increased in AML samples than in those from healthy controls.558 The inhibition of IRF3 restricted AML cell proliferation and colony formation, and promoted AML cell apoptosis, whereas overexpression of IRF3 promoted AML cell survival, which was positively correlated with the oncogenic presence of miR-155 in AML.558
A genome-wide association study (GWAS) study revealed that IRF4 was a major susceptibility gene for CLL and AML.559,560 Higher IRF4 levels attenuated BCR signaling by negatively regulating the expression of the spleen tyrosine kinase (SYK) and IKAROS in CLL cells.561 Additionally, murine CLL with low B cell-specific IRF4 expression showed a poorer prognosis due to enhanced tumor immune evasion.562 Furthermore, IRF4 expression in CD3+ T cells from CML patients was significantly reduced.563 CML patients who achieved an early molecular response had higher IRF4 values at both diagnoses and after 3 months of therapy compared to those who did not achieve early molecular response.564 In addition, PU.1 can collaborate with IRF4 and IRF8 to repress pre-B-cell leukemia.565 It was found that repression of IRF4 by miR-125b induced both myeloid and B-cell leukemia in vitro.566 However, Sun et al. demonstrated that miR-155-5p inhibited IRF4 protein degradation, thereby promoting cyclin dependent kinase 6 (CDK6) expression and facilitating childhood ALL development.97 IRF4 is also an oncogenic transcription factor in adult ATL. IRF4 gene was expressed higher in ATL cells than in normal T cells, and overexpression of IRF4 induced the upregulation of oncogenes such as MYC and baculoviral IAP repeat-containing protein 3 (BIRC3).567 Moreover, the CDK9 inhibitor alvocidib could downregulate super-enhancers-mediated IRF4, thereby inhibiting ATL cell proliferation.568
Watanabe et al. demonstrated that IRF8 expression was downregulated in CML patients, and Irf8-/- mice developed a CML-like disease.569 IRF8 partially suppressed CML development in vivo through a Fas-dependent apoptosis mechanism.570 IRF8 acts as a roadblock for β-catenin-driven leukemia. The deletion of IRF8, combined with constitutive β-catenin activation, resulted in the progression of CML into a fatal blast crisis and imatinib resistance.571 Additionally, the loss of IRF8 facilitates the initiation of acute promyelocytic leukemia (APL).572 Furthermore, IRF8 has been shown to suppress T-cell ALL proliferation and invasion by suppressing the PI3K/AKT pathway.573 However, some studies have found that high IRF8 expression was associated with poorer prognoses in AML patients, and IRF8 loss inhibited AML cell growth.574,575 Pingul et al. uncovered that the transcriptional circuit of myocyte enhancer factor 2D (MEF2D)/IRF8 was required for AML maintenance.576,577
The single-cell RNA sequencing study demonstrated that CMML-2 stem cells from CMML patients were characterized by highly expressed regulome associated with IRF1, IRF7, and IRF8, factors that are highly expressed in monocytic lineage differentiation.578 However, the detailed functions of these IRF members in CMML require further exploration.
Multiple myeloma (MM)
The roles of IRF1 and IRF3 in MM are complex. Wang et al. demonstrated that all-trans retinoic acid activated the retinoic acid receptor γ and IFN-β response pathway, which induced IRF1 overexpression to initiate 2’-5’-oligoadenylate synthetase 1 (OAS1) transcription. This caused cellular RNA degradation and cell death, enhancing MM sensitivity to carfilzomib (Cfz)-induced cytotoxicity and resensitizing Cfz-resistant MM cells to Cfz in vitro.579 Additionally, drug MEDI2228 activated the cGAS/STING/TBK1/IRF3 and STAT1/IRF1-induced IFN-I pathway to fight MM cell immune evasion.580 Moreover, macrophage inflammatory protein 1α (MIP-1α) induced osteoclast formation through activating the MEK/ERK/c-Fos pathway and suppressing the p38 map kinase (p38MAPK)/IRF3 pathway and IFN-β expression in MM.581 However, Liu et al. found that myeloma cell-secreted 2-deoxy-D-ribose induced the STAT1/IRF1 pathway activation and then upregulated CIITA expression in osteocytes to facilitate myeloma-induced bone lesions.582 In myeloma plasma cells, the interaction of ZBP1 with TBK1 and IRF3 causes IRF3 phosphorylation, which allows IRF3 to directly bind to and activate cell cycle genes, in part through cooperation with IRF4, promoting myeloma cell proliferation in MM.257
IRF4 is highly expressed in MM cells and is strictly required for MM cell survival, downregulating pro-apoptotic BCL2-modifying factor (BMF) and BCL2L11.583 Moreover, the oncogenic MAF bZIP transcription factor (MAF) activated enhancers and super-enhancers in B cells and plasma cells and cooperated with the plasma cell IRF4 to endow myeloma plasma cells with migratory and proliferative transcriptional potential.584 Overexpression of IRF4 and the oncogene c-Myc is a major mechanism of lenalidomide resistance in MM. Inhibition of SUMOylation enzymes suppressed IRF4 gene transcription and reduced IRF4 protein level by enhancing IRF4 degradation to enhance lenalidomide sensitivity.585 In addition, lenalidomide selectively degraded IKZF1, leading to IRF4 repression and IRF5 increase, which then promotes macrophage polarization from an anti-inflammatory M2-like phenotype toward a tumoricidal M1 phenotype.586 IRF4 down-regulation inhibited tumor formation and myeloma dissemination, eradicated myeloma progenitors, and improved survival and sensitivity to myeloma drugs.587,588 The bioinformatics analysis revealed that IRF7 was an important immune-related gene in MM patients, and it could promote tumor progression in vitro.589 An IRF8 mutation was reported in MM patients by the GWAS.590 In Myeloma cells, heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1) upregulated miR-92a-2-5p and miR-373-3p expression, activating osteoclastogenesis and suppressing osteoblastogenesis through inhibiting IRF8 or RUNX family transcription factor 2 (RUNX2) to drive MM osteolytic bone disease.591 These studies reveal that IRF4 and IRF7 are oncogenic, while IRF5 and IRF8 may act as tumoricidal factors in MM.
Myelodysplastic syndrome (MDS)
Although IRF1 is regarded as a tumor suppressor in pre-leukemia myelodysplasia,547 it has been reported that myelodysplasia patients without IRF1 expression had a decreased incidence of autoimmune manifestations, indicating that IRF1 may increase the probability of autoimmune phenomena in MDS, companied by a decline in quality of life.592,593 Compared to controls, MDS patients displayed upregulated expression of IRF2, IRF3, and IRF7.594 TLR3 hyperactivation can induce IRF3, IRF7, and NF-κB translocating to the nucleus, which then activates IFN-α/β transcription to promote the IFN response and result in aggressive MDS.595 The GWAS and bioinformatic analysis showed that IRF4 was a major susceptibility gene for MDS, and patients with IRF4 alterations presented worse OS than those without alterations.560,596
Head and neck SCC (HNSCC)
Wang et al. demonstrated that the inhibition of CTLA4 activated CD8+ T cells and increased IFN-γ and TNF-α levels, which in turn induced the STAT1/IRF1 axis activation to trigger tumor cell pyroptosis with tumor cell death in HNSCC cell lines.597 IRF2-induced claudin-7 upregulation to suppress oral SCC cell lines proliferation, invasion, and migration.598 The m6A demethylase alkB homolog 5, RNA demethylase (ALKBH5) overexpression inhibited RIG-I protein, resulting in the downregulation of IFN-α secretion mediated by the IKKε/TBK1/IRF3 axis. This reduction leads to reduced immune-killing cell infiltration and facilitates HNSCC progression and immune evasion.599 Yan et al. displayed that Irf6 loss triggered rapid HNSCC development in mouse models, while Notch/Ripk4/Irf6 axis activation suppressed tumor growth in vitro.600 These findings illustrate that IRF1, IRF2, IRF3, and IRF6 play an inhibitory effect on HNSCC.
Nasopharyngeal carcinoma (NPC)
IRF1 has been identified as one of the motifs enriched in the NPC unique cluster;601 however, the deeper function of IRF1 in NPC remains unclear. The IRF2 motif is also enriched in the NPC unique cluster,601 and IRF2 promotes centromere protein N (CENP-N) expression in NPC cells, further facilitates NPC cell proliferation, cell cycling, and apoptosis resistance, alongside increased aerobic glycolysis.602 Ge et al. demonstrated that circBART2.2 promoted PD-L1 expression via activating IRF3 and NF-κB in NPC, thereby inducing tumor immune escape.603 These studies suggest that IRF2 and IRF3 likely act as tumor promoters in NPC. On the other hand, Xu et al. demonstrated that IRF6 was reduced in highly metastatic NPC cells, cancer stem-like NPC cells, and NPC animal models.261 IRF6 directly restricts ATP-binding cassette sub-family G member 2 (ABCG2) expression in NPC cell lines and NPC tissues, resulting in the inhibition of NPC cancer cell proliferation, colony formation, and self-renewal.261
Cholangiocarcinoma (CCA)
IRF1 was found to be low expressed in CCA, and IRF1 acted as a tumor suppressor in regulation of CCA cell proliferation, cell cycle, migration, and invasion.604 Moreover, miR-383 could enhance proliferation, migration, and invasion of CCA cells via negatively regulating IRF1.605 Instead, IRF4 could upregulate lncRNA SOX2-OT, which further upregulated SOX2 and activated PI3K/AKT pathway to promote CCA cell proliferation and metastasis.606
Breast cancer (BC)
The potential roles of IRF1 and IRF9 in BC are controversial. IRF1 was found to be overexpressed in patients with highly aggressive BC subtypes, facilitating EMT, migration, dissemination, and metastasis formation.607 Moreover, Wu et al. showed that ubiquitin ligase E3 component N-recognition protein 5 (UBR5) enhanced PD-L1 transactivation by upregulating PKR, STAT1 and IRF1 to induce tumorigenesis in triple-negative breast cancer (TNBC).608 On the other hand, IRF1 can upregulate autophagy and apoptosis, and inhibit NF-κB activity to inhibit BC cell growth.609,610 IKKε showed its oncogenic potential by accelerating IRF1 degradation and inhibiting IRF1 transcriptional activity in a BC cell line.53 Luker et al. reported that IRF9 overexpression caused resistance to antimicrotubule agents in BC cells.611 However, Brockwell et al. displayed that IRF9 was a marker of reduced risk of distant relapse, and IRF9 loss was a poor prognostic biomarker for pre-chemotherapy in TNBC.612
Studies have revealed that IRF2, IRF3, IRF4, IRF5, IRF6, and IRF8 are protective factors in BC progression. Kriegsman et al. revealed that IRF2 negatively regulated PD-L1 expression to inhibit BC immune evasion.390 IRF3 was shown to facilitate retinoic acid (RA)/polyinosinic-polycytidylic acid (poly(I: C))-induced apoptosis in BC cells, driving the death of BC cells.613 Activated cGAS-STING-IRF3 pathway by cGAMP reversed the EMT and PI3K/AKT pathways and prevented TNBC metastasis.614 STING/TBK1/IRF3 pathway activation to recruit CD8+ T cells is a critical determinant of the therapeutic efficacy of PARP inhibition in TNBC.615 IRF4 was regarded as a tumor suppressor and higher IRF4 expression was associated with improved outcome in node-negative BC.616,617 Overexpression of IRF5 in BC cells inhibited in vitro and in vivo cell growth, and migration, and sensitized them to DNA damage. Additionally, IRF5 downregulation was correlated with increased invasiveness in human ductal carcinoma.618,619 IRF6 was downregulated in highly invasive BC cell lines, but increased in poorly aggressive ones, and drugs like bortezomib and lapatinib upregulated IRF6 expression to inhibit BC growth.620,621,622 BC-produced GSF decreased IRF8 in cDC progenitors, which was followed by reduced cDC1 and subsequently impeded immune surveillance in BC, impairing anti-tumor CD8+T-cell responses and causing poor outcomes in BC patients.462 Additionally, IRF8 deficiency negatively impacted BC therapeutic efficacy.623
Cervical cancer (CC)
The most common histological subtype of cervical cancer (CC) is SCC, followed by adenocarcinoma.624 The human papillomavirus 16 (HPV16) E6 oncoprotein facilitates CC growth and angiogenesis, while IRF1 inhibits tumourigenesis and the angiogenic activity of HPV16 E6 in CC.625 In addition, patients with overexpressed IRF1 in pretreatment CC biopsy cells had a significantly better response to neoadjuvant radio/chemotherapy, and STAT3/IRF1 pathway activation sensitizes CC cells to chemotherapeutic drugs.626 IFN-γ/IRF1 signaling also upregulates P27, which inhibits the expression and telomerase activity of human telomerase reverse transcriptase (hTERT), thereby resulting in the inhibition of CC.627 IRF4 low expression is associated with a significantly poorer OS in CC patients.628 Similarly, it has been reported that IRF6 expression was significantly downregulated in CC specimens and cell lines, and miR-587 could repress IRF6 protein expression to abrogate the antineoplastic activity of IRF6 in CC cells and promote HeLa tumor growth.629 These results indicate that the loss of IRF1, IRF4, and IRF6 expression facilitates CC development, and IRF1 pathway activation may be beneficial for enhancing the efficacy of CC chemotherapeutic drugs.
Endometrial cancer (EC)
Although Kuroboshi et al. reported that IRF1 expression was decreased in human endometrioid adenocarcinoma compared with normal endometrium and postmenopausal endometrium,630 Gao et al. revealed that enhanced IRF1 protein stability may upregulate PD-L1 in ECs and promote tumor immune escape.631 Zeng et al. presented that IL-6 enhanced reactive oxygen species (ROS) generation and induced mtDNA leakage in EC cells, which further activated the cGAS/STING/TBK1/IRF3 pathway, subsequently PD-L1 production, leading to tumor immune escape.632 The adenosine deaminase family acting on RNA1 (ADAR1) knockdown induced the expression of IRF7 and other proteins, which in turn resulted in apoptosis in EC cells.633 Therefore, IRF1 and IRF3 probably promote EC cell’s immune escape via PD-L1 production, while IRF7 may inhibit EC growth via inducing apoptosis, which is needed for further investigation.
Melanoma
NF-κB/IRF1 axis activation, in association with cDC1s, was linked to better clinical outcomes to improve cancer immunotherapy in melanoma patients.634 A bioinformatic analysis revealed that IRF1 expression might act as a biomarker indicating CD8+ T cell infiltration in melanoma.635 Compared to healthy controls, patients with metastatic melanoma had a significantly lower level of IRF1 signaling molecules in their peripheral blood lymphocytes.636 On the other hand, Yokoyama et al. revealed that SOX10 repressed IRF1 transcription via directly inducing IRF4, which then impaired PD-L1 expression in melanoma cells.637 Under ultraviolet radiation treatment, IRF3 formed a transcriptional complex with NF-κB/p65 to promote PD-L1 transcription and reduced CD8+ T-cell-mediated cytotoxicity, thereby facilitating immune evasion of oncogenic melanocytes and melanoma development.638 However, Type 2 transglutaminase negatively regulated IRF3 activation, resulting in reduced IFN-β expression and inducing immune evasion of melanoma cells.639 Moreover, Musick et al. found that IRF3 overexpression reduced tumor growth in a melanoma mouse model.640 Humblin et al. identified that IRF8 was essential for IL-9-producing Th9 cells anti-tumor effects in mouse melanoma models.641 Wang et al. illustrated that the IRF9/STAT2 signaling activation induced adaptive resistance to BRAF inhibitor therapy in melanoma by inhibiting gasdermin E (GSDME)-dependent pyroptosis in melanoma cells and in a xenograft tumor model.642 Moreover, the loss of stromal antigen 2 activated IRF9, which in turn enhanced IFN-I signaling and PD-L1 expression in melanoma.643
Osteosarcoma
Ectopic expression of IRF1 induced caspase-7 and Bcl-2 downregulation to activate apoptosis in cell lines of Ewing’s sarcoma.644 IRF1 activated miR-134 to inhibit osteosarcoma cell proliferation, invasion, and migration both in vitro and in vivo.645 Moreover, IRF1 positively regulated growth arrest specific 5 (GAS5) expression, causing upregulation of downstream tumor suppressors in osteosarcoma cell lines.646 Suppression of IRF1 expression by miR-4295 overexpression promoted osteosarcoma cell proliferation, migration, and invasion.647 Similarly, it was reported that the inhibition of IRF2 by miR-18a-5p promotes osteosarcoma cell invasion and migration.648 STING/IRF3/ IFN-β pathway activation by a sodium-glucose cotransporter 2 (SGLT2) inhibitor significantly inhibited osteosarcoma growth in vivo.649 Higher IRF7 expression tended to a better prognosis in osteosarcoma patients, and IRF7 inhibited PKM2 expression to interfere with the Warburg effect in osteosarcoma cells.650,651
Gliomas
IRF1 enhanced LGALS-9 transcription to encode galectin 9 (Gal-9) protein and then drive macrophage M2 polarization. In turn, these macrophages secreted vascular endothelial growth factor A (VEGFA) to promote angiogenesis and glioma growth.652 Overexpression of IRF2 protected glioma cells from ferroptosis and enhanced their invasive and migratory abilities in vitro.653 On the other hand, IRF3 was reported to inhibit glioma proliferation, migration, and invasion.654,655,656 Yang et al. presented that a therapeutic nanosystem (SPP-ARV-825), targeting the bromodomain and extraterminal-containing protein family 4 (BRD4)-degrading proteolytic chimera, could inhibit IRF4 promoter transcription and STAT6, STAT3, and AKT phosphorylation. This inhibition attenuated cell proliferation, induced cell apoptosis, and suppressed M2 macrophage polarization, leading to an antitumor effect in the glioma xenograft model.657 Li et al. showed that the knockdown of Gal-9 activated the TLR7/IRF5 pathway, which facilitated macrophages M1 polarization and enhanced CD8+ T cells anti-tumor effect in glioblastoma.658 IRF6 inhibited PKM2 and GLUT1 transcription to impair glycolysis and cell proliferation, and induce apoptosis in glioma cells. Furthermore, IRF6 overexpression reduced glioma xenograft tumor growth and prolonged nude mice survival.263 In addition, activation of the TLR7/MyD88/IRF5/IRF7 pathway induced IFN-β secretion, which then stimulated NK cells to eradicate glioma cells.659
IRFs and inflammatory and autoimmune diseases
Inflammatory bowel disease (IBD)
Tang et al. found that IRF1 was upregulated in human IBD and dextran sulfate sodium (DSS)-induced mice colitis.660 They also identified that TNF-α-mediated IRF1 activation suppressed osteopontin expression to inhibit p-AKT, p-P38, and p-ERK activities, and aggravate apoptosis and intestinal epithelial cell injury.660 IRF1 deficiency could restrict TNF-α-induced intestinal epithelial cells shedding to maintain intestinal barrier integrity.661 Moreover, downregulation of IRF1 expression by miR-24-3p was found to promote M2 polarization and subsequently reduced hyperinflammation-induced damage in the murine colon.662 Wang et al. showed that increased activation of IRF3 and IRF7 facilitated inflammatory chemokines expression and promoted excessive intestinal inflammation in LPS-responsive beige-like anchor-deficient mice.663 However, Chiriac et al. reported that both Irf3-/- and Irf3-/-Irf7-/- mice developed more severe colitis after DSS administration compared to control mice.664 Buchele et al. illustrated that IRF4 drove intestinal inflammation via both T cell-intrinsic and T cell-extrinsic mechanisms,665 while other studies reported that increased IRF4 expression and suppressed IRF5 phosphorylation ameliorated colitis through inducing M2 macrophage polarization.666,667 Yan et al. demonstrated that IRF5 in CD4+ T cells accelerated experimental colitis with increased chemokine migration, Th1/Th17 cytokines, and decreased Th2-associated anti-inflammatory cytokines in vivo.668 IRF5-NF-κB p65 complex formation disruption or impairment of endogenous IRF5 activation could prevent intestinal inflammation in DSS-mediated colitis.669,670 Moreover, inhibition of IRF5 by thalidomide or miR-144/451 could block M1 macrophage polarization or DC activation, which further attenuated colitis in DSS-induced models.671,672 Zhang et al. found that the adoptive transfer of CD4+T cells with IRF8 deficiency into Rag1-/-recipients enhanced colitis development, which was associated with increased gut Tfh-related gene expression, and IRF8 represses Tfh differentiation by suppressing IRF4 transcription and transactivation to prevent IBD development.673 However, Veiga et al. reported that IRF8 silencing in leukocytes presented promising anti-inflammatory properties in a murine colitis model.674
Asthma
Polymorphisms of IRF1 were reported to increase childhood allergic asthma risk, which was associated with increased pro-inflammatory gene regulation.675 IRF2 was identified as one of the key candidate genes to regulate genetic susceptibility to asthma in the Indian population.676 A study found that intracellular STING/TBK1/IRF3/7 signaling pathway activation by cGAMP exacerbated allergic inflammation and asthma.677 Moreover, IRF3 was shown to be essential for house dust mite (HDM)-induced airway allergy, and in Irf3-/- mice, HDM-mediated airway allergy were strongly attenuated when compared with those in wild-type mice.678 He et al. demonstrated that IRF7 was overexpressed in ILC2s from asthma patients compared to those from healthy controls. Furthermore, IRF7 deficiency impaired the expansion and function of lung ILC2s in multiple models of allergic asthma, leading to the remission of allergic airway inflammation.679 Studies uncovered that the activation and upregulation of IRF4 promoted Th2 cell response, M2 macrophage activation, and Th9 cell development, exacerbating allergen-induced lung allergic inflammation. Additionally, the inhibition of IRF4-IL-33 axis activation or IRF4 expression could attenuate allergic inflammation in asthma mouse models.680,681,682,683,684,685 Orissa et al. reported that IRF5 was markedly overexpressed in bronchoalveolar lavage cells of severe asthmatics compared to milder asthmatics or controls. Additionally, IRF5 drove a Th1 cell response and airway hyperreactivity in severe asthma mice.686
Psoriasis and atopic dermatitis
Based on the in silico analysis, IRF1 was reported to recognize psoriasis response elements and was a psoriasis-activated transcription factor.687 Kuai et al. revealed that IRF1 was highly expressed in human psoriasis specimens and that IRF1 inhibition alleviated psoriasis-like inflammation in vitro.688 Moreover, TNF-α inhibitors impeded STAT1- and IRF-1-independent pathways, interrupting M1 polarization and further preventing psoriasis progress.689 Studies have identified IRF2 as one of the potential susceptibility genes for psoriasis.690,691 Mice with IRF2 deficiency displayed psoriasis-like skin inflammation.692 In addition, Kawaguchi et al. illustrated that Irf2+/- mice showed more severe imiquimod (IMQ)-induced skin inflammation, with higher levels of TNF-α, IL-12/23p40, IL-17A, and IL-22, compared to normal mice.693 Moreover, IRF2 haploinsufficiency created heightened biological responses to IFN-α, leading to enhanced angiogenesis and psoriasis-like inflammation within the skin.693 Li et al. presented that the STING/IRF3 pathway was activated in palmitic acid and IMQ induced human immortalized keratinocytes (HaCaT) cells.694 Furthermore, STING and p-IRF3 expression levels were significantly increased in patients skin with psoriasis and diabetes, as well as in diabetic mice skin with psoriasis, indicating that the STING/IRF3 pathway might regulate inflammatory response in psoriasis with diabetes mellitus.694 Ni et al. illustrated that IRF4 was significantly increased in keratinocytes and inflammatory cells in psoriasis vulgaris lesions compared to normal controls.695 Cai et al. reported that IRF4 activation promoted dermal γδT cell IL-17 production,696 which indicates that IRF4 may facilitate skin inflammation in psoriasis. Additionally, Nakao et al. uncovered that IRF5 deficiency caused IRF4 upregulation in DCs, followed by IL-23 augment, inducing Th17 response amplification and exacerbating IMQ-induced psoriasis-like skin inflammation.697 The RNA sequencing revealed overexpressed IRF7 in patients with psoriatic skin.698,699 Zdhhc2 was required by pDCs to induce IRF7 phosphorylation and IFN-α production in psoriasis-like inflamed murine skin.700 Besides, TBK1 activation and IRF7 nuclear migration facilitate IMQ-induced acute psoriasis-like inflammation.701 Moreover, the bromodomain and extraterminal ___domain inhibitor NHWD-870 could inhibit IRF7 and p-IRF7 expression to ameliorate IMQ-induced psoriasis-like inflammation.699 The microarray data analysis revealed that IRF8 was an important hub gene in psoriasis complicated with atherosclerosis,702 and the RNA sequencing analysis also revealed that IRF9 was a core transcriptional regulator associated with inflammation in psoriasis.703 In cases of atopic dermatitis, Gao et al. revealed that IRF2 variants were associated with atopic dermatitis and eczema herpeticum.704 A comprehensive bioinformatics analysis reported that IRF7 was upregulated in atopic dermatitis patients and identified it as a hub gene for atopic dermatitis.705
Systemic lupus erythematosus (SLE) and related complications
Studies have uncovered that overexpressed IRF1 could produce a pattern of hyperacetylation at H4 lysine residues and induce the expression of target genes in vitro, a finding also shown in SLE patients.706,707,708,709 Moreover, IRF1 overexpression in monocytes from SLE patients enhanced inflammasome activation.710 Chen et al. reported that HDAC1 inhibited miR-124 and then promoted IRF1 expression to potentiate CD4+ T cell activation in SLE.711 In lupus nephritis (LN) patients, an inverse correlation between IRF1 and miR-130b levels was observed in renal samples. Overexpression of miR-130b in vivo suppressed IRF1 expression and consequently ameliorated IFN-α-accelerated LN.712 A joint analysis of GWAS and replication data found the missense variant in IRF3 was associated with LN patients, and identified IRF3 as a novel locus for SLE.713 Xu et al. presented that circELK4 sponged miR-27b-3p, which facilitated STING/IRF3/IFN-I signaling activation and promoted inflammation, cell apoptosis, and renal injury in LN.714 Phosphorylation of IRF3/IRF7 by TonEBP in macrophages facilitated autoimmune responses in SLE/LN pathogenesis.715 Zheng et al. found that serine/threonine kinase AKT2 interacted with IRF3 and attenuated IRF3 nuclear translocation to reduce IFN-I production, which consequently prevented SLE development.716 Additionally, Faridi et al. revealed that CD11b activation with the agonist LA1 reduced the phosphorylation of AKT and its substrate FOXO3a to suppress IRF3/7-mediated gene expression, thereby suppressing TLR and IFN-I signaling-associated inflammation and autoimmunity in SLE.717 Besides, studies showed that IRF7 was an important transcript factor in the LN process, and the lupus risk allele in IRF7 was associated with significant IRF7 hypomethylation.718,719,720 IRF7 was required for autoantibody production in murine lupus development.721 Chandrasekaran et al. illustrated that IRF4 was required for the expansion and function of effector Treg cells to limit murine lupus-like disease development.722 However, Lech et al. also demonstrated that IRF4 promoted LN development in mice.723 Genetic variants within and around IRF5 were robustly associated with SLE risk.724,725,726,727,728 IRF5 was an early regulator of human B cell activation, and monoallelic IRF5 deficiency in B cells prevented murine lupus.729,730 Moreover, recent studies revealed that both genetic loss of Irf5 and chemical inhibition ameliorated disease in murine lupus models.731,732 The IRF8 locus was also associated with an increased SLE risk.733 IRF8 was critical for the differentiation of MDSCs, which further impaired Treg differentiation and promoted Th17-cell polarization in SLE development.297,734 Studies also showed that IRF9 mRNA levels were increased in SLE monocytes, which was positively associated with both SLE activity and ISGs activity.735,736
Multiple sclerosis (MS)
Annibali et al. found IRF1 was downregulated in B cells from MS patients, and IRF1/CXCL10 axis downregulation might promote a pro-survival status of B cells in MS.737 On the other hand, Kortam et al. revealed serum IRF3 levels were upregulated in MS patients.738 Estrogen receptor alpha in DCs degraded TRAF3 via ubiquitination, reducing IRF3 nuclear translocation and transcription of membrane lymphotoxin and IFN-β components, which consequently alleviated disease severity in MS mice.739 Studies illustrated that IRF4 facilitated Th17 cells differentiation in relapsing-remitting MS,740 and miR-30a reduced IRF4 expression to inhibit Th17 differentiation and prevent the development of autoimmune encephalomyelitis (EAE) in mice, an animal model of MS.741 In addition, studies reported an IRF5 variant was associated with primary progressive MS, and variation near IRF6 was associated with IFN-β-mediated liver injury in MS.742,743 Studies also revealed that IRF8 was a risk gene for both EAE in mice and MS patients, and IRF8 SNP was associated with mitogen-activated protein kinase kinase 1 (MP2K1) phosphorylation levels, which were overactive in MS.744,745,746 IRF8 enhanced αvβ8 integrin expression in APCs and activated TGF-β signaling, resulting in Th17 cell differentiation and exacerbated neuroinflammation.747
Sjögren’s syndrome (SS) and rheumatoid arthritis (RA)
Wei et al. found that IRF4 was upregulated in murine PMN-MDSCs during experimental SS (ESS) progression in mice and IRF4 deficiency facilitated aryl hydrocarbon receptor (AhR)-induced PMN-MDSC responses to attenuate ESS.748 Furthermore, Xiao et al. presented that artesunate suppressed IRF4-mediated glycolysis and increased proteasomal degradation of IRF4 to inhibit Th17 response and ameliorate ESS in mice.749 IRF5 was a risk gene in SS.750,751 In RA, IRF1 was reported to induce IFN-β expression, which further activated the JAK/STAT pathway.752 IRF3 was reported to be strongly associated with ISG expression in RA.753 IRF4, IRF5, and IRF8 were identified as genetic risk factors for RA.754,755,756,757,758 Moreover, Duffau et al. revealed that IRF5 promoted inflammatory arthritis in an RA mouse model.759 The Fig. 9 shows the potential role of IRFs in diverse inflammatory and autoimmune diseases.

The potential role of IRFs in inflammatory and autoimmune diseases. The reported IRF family members show protective roles, negative roles, or dual roles in inflammatory bowel disease, psoriasis, systemic lupus erythematosus, and multiple sclerosis. On the other hand, the reported IRF family members almost play negative functions on atopic dermatitis, asthma, Sjögren’s syndrome and rheumatoid arthritis
IRFs and metabolic diseases
Nonalcoholic fatty liver disease (NAFLD)
NAFLD includes all kinds of fatty liver disease, from isolated hepatic steatosis or NAFL to nonalcoholic steatohepatitis (NASH) and NASH cirrhosis.760 Patel et al. revealed that hepatic IRF3 directly targets protein phosphatase 2 scaffold subunit Abeta (Ppp2r1b) to inhibit glucose production, and it was activated in obese humans with NAFLD.761 Qiao et al. revealed that STING and IRF3 levels were increased in the livers of high-fat diet fed NAFLD-like mice models.762 The STING/IRF3 pathway activation promoted hepatocyte injury, dysfunction, and liver fibrosis through inducing inflammation and apoptosis, and impeding glucose and lipid metabolism.762,763 Besides, disruption of IRF3 activation by hepatocyte nuclear factor 1 A ameliorated the innate immune response and NAFLD/NASH in vitro.764 Tong et al. demonstrated that IRF6 acted as a protective factor against liver steatosis in hepatocytes, and that hepatic IRF6 suppressed peroxisome proliferator-activated receptor γ to alleviate liver steatosis and metabolic problems in NAFLD transgenic mice.765
Alcoholic liver disease (ALD)
The IRF1-induced caspase-1 and NADPH oxidase 2 (NOX2)-mediated ROS pathway could induce a gastrin-releasing peptide receptor pro-inflammatory and oxidative stress response in alcohol-associated liver injury (ALI).766 The degradation of IRF1 and the removal of damaged mitochondria by murine macrophage autophagy prevented ALD in mice.767 Patients with alcoholic hepatitis showed elevated p-IRF3 and IRF3-mediated signals in livers, and IRF3 enhanced immune cells apoptotic death, which then resulted in hepatocellular injury in ALD mice.267 Moreover, IRF3, activated by endoplasmic reticulum stress or ethanol, also contributed to hepatocyte apoptosis and inflammatory response in early ALD.768 cGAS/IRF3 pathway expression was positively correlated with ALD severity, and hepatic gap junctions propagated cGAS-mediated IRF3 activation to enhance alcohol liver injury in ALD models.769,770
Atherosclerosis (AS)
IRF1 was overexpressed in both human and mouse AS lesions,771 and it increased CCL19 expression, facilitating vascular smooth muscle cells (VSMCs) proliferation and migration in AS.772 In addition, IRF1 facilitated non-canonical NF-κB-NLRP3-mediated endothelial pyroptosis and AS progression.773 Inhibition of IRF1 suppressed modified lipoprotein uptake, promoted cholesterol efflux, and altered gene expressionrelated to lipid metabolism to prevent AS.771 Liu et al. demonstrated that Irf3-/-Apoe-/-mice presented significantly decreased AS lesions due to suppressed VCAM-1 and intercellular adhesion molecule 1 (ICAM-1) expression, which attenuated macrophage infiltration.774 IRF5 acted as a detrimental factor in AS through maintaining pro-inflammatory macrophages, restricting necrotic core expansion o by impairing efferocytosis, and inducing rupture-prone atherosclerotic plaques formation.775,776 IRF5-deficient macrophages facilitated a stable plaque phenotype generation to combat AS in mice.777 IRF7 activation enhanced AS progress, and repression of IRF7-dependent TLR9 responses in macrophage induced decreased proatherogenic CXCL10 production.778 Interruption of the RAGE/IRF7 pathway also triggered a switch from a pro- to an anti-inflammatory environment and accelerated AS regression.779 Deficiency of IRF8 in hematopoietic cells with a CML-like phenotype was presented to promote AS progress in mice.780 However, antoher study demonstrated that DCs-specific IRF8 deletion significantly reduced aortic T-cell accumulation and adaptive immune responses, resulting in the inhibition of AS development, especially in the aortic sinus.781
Acute coronary syndrome (ACS) and myocardial ischemia reperfusion (MIR)
Guo et al. suggested that IRF1 might induce Th1 cell differentiation and thus contribute to ACS pathogenesis in vitro.782 Furthermore, in ACS patients, IRF1 was found to be overexpressed in macrophages, facilitating macrophage pyroptosis and the downstream inflammatory response in AS and ACS.783,784 The knockdown of cardiac-specific IRF1 significantly reduced infarct size, improved cardiac function, and inhibited myocardial apoptosis after MIR injury, while the overexpression of cardiac-specific IRF1 obviously enhanced MIR injury in mice.785 IRF2-driven GSDMD-dependent pyroptosis and then contributed to MI in both in vivo and vitro models.786 Activated IRF3/IFN axis in MI cardiac macrophages improved survival.787 IRF3-dependent signaling interruption led to decreased inflammatory cell infiltration, cytokines, chemokines in the heart, and recovered cardiac function.787,788 It was reported that the TLR7/MyD88/IRF5 signaling pathway activation aggravated MIR injury in mice.789 The inhibition of the Dectin1/-Syk-IRF5 pathway interrupted M1 macrophage polarization and inflammation in both in vivo and in vitro coronary microvascular dysfunction models.790 In addition, reduced IRF5 expression accelerated both cutaneous and infarct healing and alleviated post-MI heart failure development after coronary ligation.791 The sequencing indicated that IRF5 gene polymorphisms might be associated with ACS susceptibility in the Chinese population.792 IRF9 was upregulated in ischemic heart tissue after MIR injury, and IRF9 ablation prevented MIR-mediated cardiomyocyte inflammation, death, and heart dysfunction.793
Obesity and diabetes
Friesen et al. demonstrated that IRF1 expression in adipocytes contributed to the upregulating of obesity-related inflammatory processes and metabolic dysregulation both in vitro and in vivo.794 IRF3 expression was increased in the adipocytes of obese mice and humans. TLR3/4-IRF3 activation induced insulin resistance in murine adipocytes, while IRF3 knockdown prevented insulin resistance. This indicates that IRF3 facilitates adipose inflammation and insulin resistance and supresses browning.795 In addition, IRF3 is a strong repressor of adipose thermogenesis via ISG15-mediated reprogramming of glycolysis.796 IRF3 directly transcriptional regulates glucose homeostasis through induction of Ppp2r1b, and subsequent suppression of glucose production.761 Moreover, STING/IRF3 axis triggered endothelial inflammation in response to free fatty acid-mediated mitochondrial damage in diet-induced obesity.797,798 Furthermore, STING/IRF3 pathway activation triggered pancreatic β cells inflammation and apoptosis, causing β-cell damage and dysfunction, while STING or IRF3 silencing ameliorated inflammation and apoptosis, and reversed impaired insulin synthesis in type 2 diabetes.799 Eguchi et al. reported that mice with a myeloid cell-specific IRF4 knockout developed serious insulin resistance on a high-fat diet. Furthermore, Irf4-/- adipose tissue macrophages enhanced M1 polarization,800 indicating that IRF4 negatively regulates inflammation in diet-induced obesity. Cavallari et al. also showed IRF4 was responsible for NOD2-induced insulin sensitizing and anti-inflammatory effects during obesity and endotoxemia.801 Studies have identified that IRF5 gene expression is positively associated with adipose inflammatory signatures in both obesity and diabetic obese patients.802,803 IRF5 facilitated M1 macrophage polarization and regulated mitochondrial architecture remodeling in obesity.101,804 Moreover, Irf5 deficiency in macrophages promoted beneficial adipose tissue expansion and insulin sensitivity during obesity in vivo.805 Studies demonstrated that suppressing IRF7 expression restored mtRNA-induced mitobiogenesis and thermogenesis, improved glucose and lipid homeostasis and insulin sensitivity, and finally mitigated obesity.806,807 Additionally, GSDMD interacted with IRF7 and subsequently formed a complex to promote adipocyte pyroptosis.808 IRF7-STAT2 cascade activation facilitated IFN-α signaling expression in islets and amplified the process of type 1 diabetes.809 Wang et al. reported that IRF9 was more downregulated in the livers of obese mice than in controls. Hepatic IRF9 overexpression in obese mice significantly attenuated hepatic steatosis and inflammation, and improved hepatic insulin sensitivity.810 The Fig. 10 presents the potential role of IRFs in diverse metabolic diseases.

The potential role of IRFs in metabolic and other diseases. a The roles of IRFs in metabolic diseases. b The roles of IRFs in other diseases. In brief, IRF1, IRF2, IRF5, and IRF7 present negative roles in these diseases. IRF3, IRF4, and IRF8 play protective roles, negative roles, or dual roles in these diseases. IRF6 is protective in non-alcoholic fatty liver disease, while a negative factor in kidney injury. IRF9 plays negative role in acute coronary syndrome and myocardial ischemia reperfusion, and kidney injury, while IRF9 plays protective role in obesity and diabetes
IRFs and other diseases
There are several studies uncovered the possible relationships between IRFs and other kinds of human diseases involved in the heart, lungs, kidneys, and mental and nervous systems (Fig. 10).
Cardiomyopathy
IRF1 elicited cardiac prohypertrophic effects by directly activating iNOS.811 Studies illustrated that the activated cGAS-STING pathway and downstream targets like IRF3 overexpression induced cardiomyocyte inflammation, apoptosis, and pyroptosis in diabetic cardiomyopathy (DCM) mouse model.812,813 Xie et al. showed that IRF3 suppression by ZNF593-AS ameliorated cardiac cell death and inflammation in DCM.814 Additionally, STING/IRF3 axis activation could activate NLRP3 and exert apoptosis, pyroptosis, and inflammation in sepsis and sepsis-induced cardiomyopathy (SIC) models.815 Gonzalez et al. identified IRF7 as the main mediator of autoinflammatory responses caused by ADAR1 absence in cardiomyocytes, resulting in late-onset autoinflammatory myocarditis progressing into DCM.816 IRF8 was found to inhibit calcineurin signaling and thus inhibit the hypertrophic response.48
Acute Lung Injury (ALI) and Acute respiratory distress syndrome (ARDS)
Wu et al. uncovered that IRF1 facilitated caspase1-mediated pyroptosis in AMs and induced downstream inflammatory cytokine release. IRF1 knockout mice significantly abrogated pyroptosis in AMs and attenuated LPS-induced lung injury and systemic inflammation.817 Moreover, IRF1 deficiency strongly alleviated neutrophil extracellular traps (NETs) generation and ROS production in neutrophils from bronchoalveolar lavage fluid.818 Chen et al. showed that endothelial-specific Irf1 knockout in ALI mice presented lung endothelium regeneration. Irf1 transactivation by LPS-induced Stat1Ser727 phosphorylation upregulated leukemia inhibitory factor (Lif), indicating that p-Stat1/Ser727/Irf1/Lif axis inhibited lung endothelial cell regeneration post-LPS injury, and IRF1 or LIF inhibition might be a promising option for improving endothelial cell regeneration and clinical outcomes in ARDS patients.819 Wang et al. presented that the STING/IRF3 pathway activation contributed to paraquat-induced ALI.820 In addition, the TBK1/IRF3 and other related pathways activation by the STING agonist diABZI amplified the inflammatory loop in ARDS.821 Zhang et al. found that IFIH1 was strongly connected with ARDS, and IFIH1 could activate IRF3 to promote M1 polarization in vitro.822 Anisodamine was shown to inhibit G9a-mediated methylation and IRF4 expression, promoting macrophage M2 polarization and inhibiting M1 polarization, which then attenuated LPS-induced ALI and pulmonary edema.823 IRF5 inhibition induced M2 polarization to improve ALI/ARDS in vitro.824 A bioinformatics analysis identified that IRF7 might regulate critical processes in ALI pathogenesis.825 Furthermore, reduced IRF7 levels or activity in the lungs of mice led to decreased IFNα mRNA levels, reduced neutrophil infiltration in the lungs, and prolonged survival of IAV-induced ALI mice.826
Kidney injury and kidney-related diseases
IRF1 was found to cause ROS-mediated IFN-α production in murine ischemic acute kidney injury (AKI) and was an important early pro-inflammatory signal during ischemic AKI in vitro and in vivo.827,828 Yan et al. illustrated that lncRNA NEAT1 inhibited miR-130a-3p to upregulate IRF1 signaling and activate TLR4/NF-κB pathways, facilitating oxidative damage during calcium oxalate (CaOx) crystal deposition.829 Moreover, studies uncovered that inhibition of IRF1 with HIF-1α or TLR4 expression by AhR or sulforaphane resulted in M1 polarization inhibition and M2 polarization progress, and finally alleviated renal CaOx crystal deposition and nephrocalcinosis-induced kidney inflammation and injury in vitro and in vivo.830,831,832 IRF1 was also found to be overexpressed in fibrotic kidney of chronic kidney disease (CKD) patients and was identified as a driver for fibrosis, which repressed Klotho expression to promote renal fibrosis in vitro and in vivo.833,834 A GWAS identified SNPs near IRF2 were associated with AKI,835 while Renken et al. reported that there was no association between genetic loci near IRF2 and AKI in the critically Ill.836 Zhang et al. demonstrated that IRF2 was increased in the serum of sepsis-mediated AKI patients and LPS-induced HK-2 cells. IRF2 downregulation induced cell proliferation and inhibited cell death, and IRF2 knockdown inhibited LPS-treated HK-2 cell pyroptosis by upregulating the expression of caspase-4, caspase-11, and GSDMD. In the caecal ligation and puncture (CLP)-induced Irf2-/- animal models, the survival rate was increased, and pathological features and scores were alleviated.837 Similarly, He et al. showed that IRF2 knockdown played anti-inflammatory and antioxidant stress functions to further decrease LPS-induced renal tissue injury in vivo and in vitro.838 IRF3 activation triggered the Hippo pathway and then impeded proliferation and repair in surviving renal tubular epithelial cells, and exacerbated LPS-induced AKI progress in vitro cell model.839 Overexpressed TRIM3 inhibited the IRF3 pathway and NLRP3 inflammasome activation to prevent LPS-induced AKI in rat models.840 Moreover, STING/TBK1/IRF3/NF-κB signaling activation in dsDNA-induced Aim2-deficient macrophages aggravated inflammatory phenotypes in a rhabdomyolysis-induced AKI mouse model.841 A genome-wide meta-analyses showed that IRF4 risk loci participated in membranous nephropathy and IgA nephropathy (IgAN) pathogenesis.842,843 IRF4 was overexpressed and was a hub gene in drug-induced AKI cell and mouse models, as well as in human specimens based on integrated transcriptomic analysis.844 Irf4 deletion in myeloid cells inhibited AKT-related monocyte recruitment to the injured kidney, and decreased activation and subsequent profibrotic M2 polarization to protect against tubulointerstitial fibrosis development after severe ischemia-reperfusion injury (IRI) in mice.845 Similarly, other studies also found that inhibition of IRF4 disrupted macrophage to myofibroblast differentiation in the kidneys under a folic acid (FA)-induced AKI-CKD transition mouse model, and protected mice against kidney inflammation and fibrosis in deoxycorticosterone acetate/salt hypertension.846,847 However, Lorenz et al. showed that IRF4 prevented CKD and kidney fibrosis following IRI, and Irf4–/– mice presented chronic intrarenal inflammation, tubular epithelial cell loss, and renal fibrosis after IRI.847,848 Liu et al. reported that ENSMUST_147219 sponged miR-221-5p to upregulate IRF6 expression to drive apoptosis and promote ischemic AKI development.849 IRF8 was a risk locus that caused abnormal IgA levels and a high polygenic score for IgAN was associated with an earlier onset of kidney failure.850 IRF8 upregulated renal tubular cell apoptosis in cisplatin-induced AKI, and that hypermethylation in the Irf8 promoter region, which repressed Irf8, could protect against cisplatin-induced AKI mice.851 However, Li et al. identified that IRF8 was indispensable for kidney cDC1s development, and that IRF8-mediated cDC1s mildly protect mice against post-ischemic AKI/AKD.852 The study by Liu et al. showed that IRF9 downregulated SIRT1 expression and increased acetylated p53 to promote rat renal cell apoptosis in hyperlipidemia acute pancreatitis associated with KI.853
Mental and nervous system diseases
IRF1 was associated with an increased risk of the development of depression.854 IRF3 was reported to be critical in maintaining the balance between neuronal excitation and inhibition, and a lack of IRF3-induced anxiety/depression-like behaviors in mice.855 IRF3 and IRF7 were identified as risk candidate genes for schizophrenia.856,857 Xu et al. uncovered that cGAMP/STING/IRF3 pathway stimulation induced triggering receptor expressed on myeloid cells 2 (TREM2) expression, which further decreased amyloid-β (Aβ) deposition and neuron loss to improve Alzheimer’s disease (AD) pathomorphology and cognitive impairment.858 However, Guo et al. showed that IRF3 expression and phosphorylation were significantly elevated during the development of AD, and the silencing of ZBP1 could suppress IRF3 to inhibit cell injury and pyroptosis of neurons, thereby improving cognitive function in rats with AD.859 Additionally, silencing IRF3 alleviated chronic neuropathic pain after chronic constriction injury by inhibiting NF-κB signaling activation in rats.860 Studies showed that IRF8 drove the expression of microglial markers linked to AD, and IRF8 overexpression exacerbated microglial activation and neuroinflammation in AD.861,862
Concluding remarks and future perspectives
This review provides a thorough examination of the structure, post-transcriptional modification sites, functional roles, and signaling pathways involving nine IRF family members across diverse cells, tissues, and human diseases. IRF family members exhibit multifaceted functions with certain activities being mutually synergistic or antagonistic. Additionally, IRF family members influence various signaling pathways differently across distinct cell types. Moreover, IRFs play complex and varied roles in immune cells, stem cells, tumor cells, and in the context of inflammatory and autoimmune diseases by engaging in numerous signaling pathways, with their regulation varying based on cell type, cellular environment, and interactions between IRF members. In immune cells, IRF family members are pivotal in the regulation of immune responses, primarily through their participation in the IFN signaling pathway. During viral infections, virus recognition receptors activate IRF3 and IRF7, which initiates IFN-I expression and the antiviral immune response. IRF4 is particularly critical for Th cell differentiation, while IRF8 plays a critical role in the maturation of bone marrow cells. In some cases, IRF1 can induce apoptosis and inhibit tumor cell growth by promoting the expression of apoptosis-associated genes, while IRF2 can promote cell survival and tumor proliferation by inhibiting the activity of IRF1. IRF5 is important in orchestrating the inflammatory response and in triggering autoimmune diseases via the activation of pro-inflammatory cytokines like IL-6 and TNF-α. In concert with STAT1 and STAT2, IRF9 forms the ISGF3 complex, which is involved in the IFN signaling pathway, regulating gene expression essential for antiviral defense and the control of autoimmune diseases.
In summary, IRF proteins serve critical functions in both human health and disease, attributed to their extensive engagement in a variety of physiological and pathological mechanisms. Research indicates that IRF family members are expressed aberrantly in an array of diseases, notably infections, inflammatory conditions, and a spectrum of cancers, affecting numerous systems including but not limited to the cardiovascular, pulmonary, urinary, reproductive, and integumentary systems. Depending on the cell types and the surrounding environment, IRF proteins can exert markedly different regulatory effects, rendering their roles intricate, and they may as double-edged swords in human health. For example, in infectious diseases, nearly all IRF members display protective functions that combat pathogens. However, in certain scenarios, activation of IRFs can trigger an inflammatory response, leading to damage within the body. Similarly, in the context of cancer, certain IRF members function as tumor suppressors, while others may behave as oncoproteins. Besides, there are IRF members that not only enhance antitumor responses but also aid in cancer immune evasion. The reasons for these discrepancies remain elusive and need further in-depth research.
Moreover, IRF proteins may function as biomarkers and therapeutic targets for diverse human diseases. Mouse models with knockout of IRF family genes exhibit a diverse array of phenotypes affecting immune system development and function, antiviral and anti-tumor immune responses, as well as cell differentiation and development (Table 1). Investigating these phenotypes provides valuable insights for identifying potential therapeutic targets for various diseases. Although therapeutic strategies on IRFs are mainly limited to indirect modulation, several direct inhibition and activated strategies have been reported in recent years (Table 2), which hold immunotherapeutic potential in the treatment of infections, inflammatory conditions, and tumors. More clinical researches are needed to explore their therapeutic potential. Therapies that augment IRF activity may be advantageous in combating a wide spectrum of pathogens; however, they could also pose significant risks to the host if uncontrolled immune activation leads to autoimmune-like disease. Considering the diverse roles played by IRF family members, unraveling the intricate mechanisms that underpin both their protective and deleterious effects is anticipated to yield valuable insights into the biology of microbial infections and host defense, autoimmune and inflammatory diseases, as well as cancers.
References
Tamura, T., Yanai, H., Savitsky, D. & Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 (2008).
Santana-de Anda, K., Gómez-Martín, D., Díaz-Zamudio, M. & Alcocer-Varela, J. Interferon regulatory factors: beyond the antiviral response and their link to the development of autoimmune pathology. Autoimmun. Rev. 11, 98–103 (2011).
Lohoff, M. & Mak, T. W. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat. Rev. Immunol. 5, 125–135 (2005).
Stoy, N. Involvement of Interleukin-1 Receptor-Associated Kinase 4 and Interferon Regulatory Factor 5 in the Immunopathogenesis of SARS-CoV-2 Infection: Implications for the Treatment of COVID-19. Front. Immunol. 12, 638446 (2021).
Jensen, M. A. & Niewold, T. B. Interferon regulatory factors: Critical mediators of human lupus. Transl. Res. 165, 283–295 (2015).
Salem, S., Salem, D. & Gros, P. Role of IRF8 in immune cells functions, protection against infections, and susceptibility to inflammatory diseases. Hum. Genet. 139, 707–721 (2020).
Zhang, Y. & Li, H. Reprogramming interferon regulatory factor signaling in cardiometabolic diseases. Physiol. (Bethesda) 32, 210–223 (2017).
Lukhele, S. et al. The transcription factor IRF2 drives interferon-mediated CD8(+) T cell exhaustion to restrict anti-tumor immunity. Immunity 55, 2369–2385.e2310 (2022).
Alfarano, G., Audano, M. & Di Chiaro, P. Interferon regulatory factor 1 (IRF1) controls the metabolic programmes of low-grade pancreatic cancer cells. Gut 72, 109–128 (2023).
Nguyen, H., Hiscott, J. & Pitha, P. M. The growing family of interferon regulatory factors. Cytokine Growth Factor. Rev. 8, 293–312 (1997).
Nehyba, J., Hrdlicková, R., Burnside, J. & Bose, H. R. Jr A novel interferon regulatory factor (IRF), IRF-10, has a unique role in immune defense and is induced by the v-Rel oncoprotein. Mol. Cell. Biol. 22, 3942–3957 (2002).
Zhao, X. et al. Characterization of DNA binding and nuclear retention identifies zebrafish IRF11 as a positive regulator of IFN antiviral response. J. Immunol. 205, 237–250 (2020).
Fujita, T. et al. Evidence for a nuclear factor(s), IRF-1, mediating induction and silencing properties to human IFN-beta gene regulatory elements. EMBO. J. 7, 3397–3405 (1988).
Miyamoto, M. et al. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell 54, 903–913 (1988).
Harada, H. et al. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 58, 729–739 (1989).
Driggers, P. H. et al. An interferon gamma-regulated protein that binds the interferon-inducible enhancer element of major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. USA 87, 3743–3747 (1990).
Nelson, N., Marks, M. S., Driggers, P. H. & Ozato, K. Interferon consensus sequence-binding protein, a member of the interferon regulatory factor family, suppresses interferon-induced gene transcription. Mol. Cell. Biol. 13, 588–599 (1993).
Fu, X. Y. et al. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. USA 87, 8555–8559 (1990).
Kessler, D. S., Veals, S. A., Fu, X. Y. & Levy, D. E. Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes. Dev. 4, 1753–1765 (1990).
Fu, X. Y. et al. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc. Natl. Acad. Sci. USA 89, 7840–7843 (1992).
Au, W. C. et al. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl Acad. Sci. USA 92, 11657–11661 (1995).
Matsuyama, T. et al. Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE). Nucleic Acids Res. 23, 2127–2136 (1995).
Mittrücker, H. W. et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275, 540–543 (1997).
Eisenbeis, C. F., Singh, H. & Storb, U. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes. Dev. 9, 1377–1387 (1995).
Yamagata, T. et al. A novel interferon regulatory factor family transcription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of interferon-regulated genes. Mol. Cell. Biol. 16, 1283–1294 (1996).
Marecki, S. & Fenton, M. J. The role of IRF-4 in transcriptional regulation. J. Interferon Cytokine Res. 22, 121–133 (2002).
Taniguchi, T., Ogasawara, K., Takaoka, A. & Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655 (2001).
Barnes, B. J., Moore, P. A. & Pitha, P. M. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J. Biol. Chem. 276, 23382–23390 (2001).
Schutte, B. C. et al. Microdeletions at chromosome bands 1q32-q41 as a cause of Van der Woude syndrome. Am. J. Med. Genet. 84, 145–150 (1999).
Kumaran, S., Dogra, S. & Kanwar, A. J. Van der Woude syndrome. Clin. Exp. Dermatol. 29, 434 (2004).
Zhang, L. & Pagano, J. S. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 17, 5748–5757 (1997).
Zhou, H., Tang, Y. D. & Zheng, C. Revisiting IRF1-mediated antiviral innate immunity. Cytokine Growth Factor. Rev. 64, 1–6 (2022).
Trujillo-Ochoa, J. L., Kazemian, M. & Afzali, B. The role of transcription factors in shaping regulatory T cell identity. Nat. Rev. Immunol. 23, 842–856 (2023).
Roberts, B. K., Collado, G. & Barnes, B. J. Role of interferon regulatory factor 5 (IRF5) in tumor progression: Prognostic and therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 1879, 189061 (2024).
Fujita, T. et al. Induction of the transcription factor IRF-1 and interferon-beta mRNAs by cytokines and activators of second-messenger pathways. Proc. Natl Acad. Sci. Usa. 86, 9936–9940 (1989).
Au, W. C. et al. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J. Biol. Chem. 273, 29210–29217 (1998).
Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 282, 15325–15329 (2007).
Lin, R., Mamane, Y. & Hiscott, J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275, 34320–34327 (2000).
Zhang, L., Wu, L., Hong, K. & Pagano, J. S. Intracellular signaling molecules activated by Epstein-Barr virus for induction of interferon regulatory factor 7. J. Virol. 75, 12393–12401 (2001).
Zhang, L. & Pagano, J. S. Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. J. Virol. 74, 1061–1068 (2000).
Kim, T. K. et al. Chemotherapeutic DNA-damaging drugs activate interferon regulatory factor-7 by the mitogen-activated protein kinase kinase-4-cJun NH2-terminal kinase pathway. Cancer Res. 60, 1153–1156 (2000).
Zhang, L. & Pagano, J. S. Interferon regulatory factor 7 mediates activation of Tap-2 by Epstein-Barr virus latent membrane protein 1. J. Virol. 75, 341–350 (2001).
Shindo, H. et al. Interferon regulatory factor-4 activates IL-2 and IL-4 promoters in cooperation with c-Rel. Cytokine 56, 564–572 (2011).
De Silva, N. S., Simonetti, G., Heise, N. & Klein, U. The diverse roles of IRF4 in late germinal center B-cell differentiation. Immunol. Rev. 247, 73–92 (2012).
Xu, W. D., Pan, H. F., Ye, D. Q. & Xu, Y. Targeting IRF4 in autoimmune diseases. Autoimmun. Rev. 11, 918–924 (2012).
Kesper, C. et al. Impact of the transcription factor IRF8 on limbal epithelial progenitor cells in a mouse model. Exp. Eye. Res. 218, 108985 (2022).
Li, W. et al. Interferon consensus sequence-binding protein is constitutively expressed and differentially regulated in the ocular lens. J. Biol. Chem. 274, 9686–9691 (1999).
Jiang, D. S. et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat. Commun. 5, 3303 (2014).
Mancl, M. E. et al. Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms. Multiple isoforms with distinct cell type-specific expression, localization, regulation, and function. J. Biol. Chem. 280, 21078–21090 (2005).
Gothe, F. et al. Aberrant inflammatory responses to type I interferon in STAT2 or IRF9 deficiency. J. Allergy Clin. Immunol. 150, 955–964.e916 (2022).
Deribe, Y. L., Pawson, T. & Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 (2010).
Garvin, A. J. et al. GSK3β-SCFFBXW7α mediated phosphorylation and ubiquitination of IRF1 are required for its transcription-dependent turnover. Nucleic Acids Res 47, 4476–4494 (2019).
Remoli, A. L. et al. IκB kinase-ε-mediated phosphorylation triggers IRF-1 degradation in breast cancer cells. Neoplasia 22, 459–469 (2020).
Kautz, B., Kakar, R., David, E. & Eklund, E. A. SHP1 protein-tyrosine phosphatase inhibits gp91PHOX and p67PHOX expression by inhibiting interaction of PU.1, IRF1, interferon consensus sequence-binding protein, and CREB-binding protein with homologous Cis elements in the CYBB and NCF2 genes. J. Biol. Chem. 276, 37868–37878 (2001).
Lin, R. & Hiscott, J. A role for casein kinase II phosphorylation in the regulation of IRF-1 transcriptional activity. Mol. Cell. Biochem. 191, 169–180 (1999).
Lin, R., Heylbroeck, C., Pitha, P. M. & Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996 (1998).
Sharma, S. et al. Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 (2003).
Hemmi, H. et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650 (2004).
Fitzgerald, K. A. et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003).
Lin, R., Mamane, Y. & Hiscott, J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 19, 2465–2474 (1999).
Xiao, J. et al. Targeting 7-dehydrocholesterol reductase integrates cholesterol metabolism and IRF3 activation to eliminate infection. Immunity 52, 109–122.e106 (2020).
Zhang, B. et al. The TAK1-JNK cascade is required for IRF3 function in the innate immune response. Cell. Res. 19, 412–428 (2009).
Xu, J. et al. IRF3-binding lncRNA-ISIR strengthens interferon production in viral infection and autoinflammation. Cell. Rep. 37, 109926 (2021).
Wang, S. et al. LPA maintains innate antiviral immunity in a pro-active state via STK38L-mediated IRF3 Ser303 phosphorylation. Cell. Rep. 41, 111661 (2022).
Meng, F. et al. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes. Dev. 30, 1086–1100 (2016).
Wang, L. et al. LMP1 signaling pathway activates IRF4 in latent EBV infection and a positive circuit between PI3K and Src is required. Oncogene 36, 2265–2274 (2017).
Biswas, P. S. et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Invest. 120, 3280–3295 (2010).
Ngwa, C. et al. Phosphorylation of microglial IRF5 and IRF4 by IRAK4 regulates inflammatory responses to ischemia. Cells 10, 276 (2021).
Ren, J., Chen, X. & Chen, Z. J. IKKβ is an IRF5 kinase that instigates inflammation. Proc. Natl. Acad. Sci. USA 111, 17438–17443 (2014).
Chang Foreman, H. C., Van Scoy, S., Cheng, T. F. & Reich, N. C. Activation of interferon regulatory factor 5 by site specific phosphorylation. PLoS. One 7, e33098 (2012).
Lopez-Pelaez, M. et al. Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc. Natl Acad. Sci. Usa. 111, 17432–17437 (2014).
Balkhi, M. Y., Fitzgerald, K. A. & Pitha, P. M. IKKalpha negatively regulates IRF-5 function in a MyD88-TRAF6 pathway. Cell. Signal. 22, 117–127 (2010).
Oberbeck, N. et al. The RIPK4-IRF6 signalling axis safeguards epidermal differentiation and barrier function. Nature 574, 249–253 (2019).
Kwa, M. Q. et al. Receptor-interacting protein kinase 4 and interferon regulatory factor 6 function as a signaling axis to regulate keratinocyte differentiation. J. Biol. Chem. 289, 31077–31087 (2014).
Uematsu, S. et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 201, 915–923 (2005).
Hoshino, K. et al. IkappaB kinase-alpha is critical for interferon-alpha production induced by toll-like receptors 7 and 9. Nature 440, 949–953 (2006).
Pfaller, C. K. & Conzelmann, K. K. Measles virus V protein is a decoy substrate for IkappaB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J. Virol. 82, 12365–12373 (2008).
Wang, L. et al. Protein phosphatase 1 abrogates IRF7-mediated type I IFN response in antiviral immunity. Eur. J. Immunol. 46, 2409–2419 (2016).
Smith, E. J. et al. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J. Biol. Chem. 276, 8951–8957 (2001).
Lee, K. J., Lee, H. & Joo, C. H. Negative Regulation of IKKε-Mediated IRF7 Phosphorylation by HSP70. J. Immunol. 204, 2562–2574 (2020).
Liang, Q. et al. ORF45 of Kaposi’s sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKε and TBK1 as an alternative substrate. J. Virol. 86, 10162–10172 (2012).
Liu, B. Q., Jin, J. & Li, Y. Y. Ubiquitination modification: critical regulation of IRF family stability and activity. Sci. China Life. Sci. 64, 957–965 (2021).
Popovic, D., Vucic, D. & Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253 (2014).
Pion, E., Narayan, V., Eckert, M. & Ball, K. L. Role of the IRF-1 enhancer ___domain in signalling polyubiquitination and degradation. Cell. Signal. 21, 1479–1487 (2009).
Nakagawa, K. & Yokosawa, H. Degradation of transcription factor IRF-1 by the ubiquitin-proteasome pathway. The C-terminal region governs the protein stability. Eur. J. Biochem. 267, 1680–1686 (2000).
Remoli, A. L. et al. HIV-1 tat recruits HDM2 E3 ligase to target IRF-1 for ubiquitination and proteasomal degradation. mBio 7, e01528-16 (2016).
Tulli, L. et al. Src family kinases regulate interferon regulatory factor 1 K63 ubiquitination following activation by TLR7/8 vaccine adjuvant in human monocytes and B cells. Front. Immunol. 9, 330 (2018).
Harikumar, K. B. et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat. Immunol. 15, 231–238 (2014).
Wang, Y. et al. African swine fever virus MGF360-14L negatively regulates type I interferon signaling by targeting IRF3. Front. Cell. Infect. Microbiol. 11, 818969 (2021).
Zhang, M. et al. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell. Res. 18, 1096–1104 (2008).
Chen, X. et al. Ubiquitin E3 ligase MID1 inhibits the innate immune response by ubiquitinating IRF3. Immunology 163, 278–292 (2021).
Zhang, W. et al. JMJD6 negatively regulates cytosolic RNA induced antiviral signaling by recruiting RNF5 to promote activated IRF3 K48 ubiquitination. PLoS. Pathog. 17, e1009366 (2021).
Ran, Y. et al. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell. Biol. 3, 283–292 (2011).
Chattopadhyay, S. et al. Ubiquitination of the transcription factor IRF-3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 44, 1151–1161 (2016).
Raja, R. & Sen, G. C. The antiviral action of the RIG-I induced pathway of apoptosis (RIPA) is enhanced by its ability to degrade Otulin, which deubiquitinates IRF3. Cell. Death. Differ. 29, 504–513 (2022).
Wang, J. et al. RNF2 promotes the progression of colon cancer by regulating ubiquitination and degradation of IRF4. Biochim. Biophys. Acta Mol. Cell. Res. 1869, 119162 (2022).
Sun, X. et al. microRNA-155-5p initiates childhood acute lymphoblastic leukemia by regulating the IRF4/CDK6/CBL axis. Lab. Invest. 102, 411–421 (2022).
Li, X. et al. Cbl ubiquitin ligases control B cell exit from the germinal-center reaction. Immunity 48, 530–541.e536 (2018).
Guo, Z. et al. Ubiquitin specific peptidase 4 stabilizes interferon regulatory factor protein and promotes its function to facilitate interleukin-4 expression in T helper type 2 cells. Int. J. Mol. Med. 40, 979–986 (2017).
Balkhi, M. Y., Fitzgerald, K. A. & Pitha, P. M. Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Mol. Cell. Biol. 28, 7296–7308 (2008).
Kim, D. et al. Cytosolic pellino-1-mediated K63-linked ubiquitination of IRF5 in M1 macrophages regulates glucose intolerance in obesity. Cell. Rep. 20, 832–845 (2017).
Chen, H. et al. Peli1 deletion in macrophages attenuates myocardial ischemia/reperfusion injury by suppressing M1 polarization. J. Leukoc. Biol. 113, 95–108 (2023).
Yu, Y., Wang, S. E. & Hayward, G. S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70 (2005).
Yu, Y. & Hayward, G. S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877 (2010).
Ning, S. & Pagano, J. S. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 84, 6130–6138 (2010).
Wang, J. et al. Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J. Immunol. 191, 3393–3399 (2013).
Young, J. A. et al. Fas-associated death ___domain (FADD) and the E3 ubiquitin-protein ligase TRIM21 interact to negatively regulate virus-induced interferon production. J. Biol. Chem. 286, 6521–6531 (2011).
Huye, L. E., Ning, S., Kelliher, M. & Pagano, J. S. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell. Biol. 27, 2910–2918 (2007).
Ning, S. et al. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28, 6536–6546 (2008).
Ling, T. et al. TARBP2 inhibits IRF7 activation by suppressing TRAF6-mediated K63-linked ubiquitination of IRF7. Mol. Immunol. 109, 116–125 (2019).
Xiong, H. et al. Ubiquitin-dependent degradation of interferon regulatory factor-8 mediated by Cbl down-regulates interleukin-12 expression. J. Biol. Chem. 280, 23531–23539 (2005).
Kong, H. J. et al. Cutting edge: autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF-8 and enhances cytokine expression in macrophages. J. Immunol. 179, 26–30 (2007).
Lin, R. et al. USP4 interacts and positively regulates IRF8 function via K48-linked deubiquitination in regulatory T cells. FEBS Lett. 591, 1677–1686 (2017).
Zhang, M. et al. Herpes simplex virus type 2 inhibits type I IFN signaling mediated by the novel E3 ubiquitin protein ligase activity of viral protein ICP22. J. Immunol. 205, 1281–1292 (2020).
Vertegaal, A. C. O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell. Biol. 23, 715–731 (2022).
Liang, Y. C. et al. SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 6, 26509 (2016).
Bergink, S. & Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458, 461–467 (2009).
Meulmeester, E. & Melchior, F. Cell biology: SUMO. Nature 452, 709–711 (2008).
Kunz, K., Piller, T. & Müller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell. Sci. 131, jcs211904 (2018).
Nakagawa, K. & Yokosawa, H. PIAS3 induces SUMO-1 modification and transcriptional repression of IRF-1. FEBS Lett. 530, 204–208 (2002).
Kim, E. J., Park, J. S. & Um, S. J. Ubc9-mediated sumoylation leads to transcriptional repression of IRF-1. Biochem. Biophys. Res. Commun. 377, 952–956 (2008).
Park, S. M. et al. SUMOylated IRF-1 shows oncogenic potential by mimicking IRF-2. Biochem. Biophys. Res. Commun. 391, 926–930 (2010).
Park, J. et al. Elevated level of SUMOylated IRF-1 in tumor cells interferes with IRF-1-mediated apoptosis. Proc. Natl. Acad. Sci. USA 104, 17028–17033 (2007).
Sun, T. et al. α-Lipoic acid (α-LA) inhibits the transcriptional activity of interferon regulatory factor 1 (IRF-1) via SUMOylation. Toxicol. Vitr. 28, 1242–1248 (2014).
Jeong, H. Y. et al. 5-Azacytidine modulates interferon regulatory factor 1 in macrophages to exert a cardioprotective effect. Sci. Rep. 5, 15768 (2015).
Han, K. J., Jiang, L. & Shu, H. B. Regulation of IRF2 transcriptional activity by its sumoylation. Biochem. Biophys. Res. Commun. 372, 772–778 (2008).
Kubota, T. et al. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 283, 25660–25670 (2008).
Maarifi, G. et al. MxA mediates SUMO-induced resistance to vesicular stomatitis virus. J. Virol. 90, 6598–6610 (2016).
Chang, T. H. et al. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS. Pathog. 5, e1000493 (2009).
Bentz, G. L., Shackelford, J. & Pagano, J. S. Epstein-Barr virus latent membrane protein 1 regulates the function of interferon regulatory factor 7 by inducing its sumoylation. J. Virol. 86, 12251–12261 (2012).
Liang, Q. et al. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187, 4754–4763 (2011).
Wang, F. et al. Loss of ubiquitin-conjugating enzyme E2 (Ubc9) in macrophages exacerbates multiple low-dose streptozotocin-induced diabetes by attenuating M2 macrophage polarization. Cell. Death. Dis. 10, 892 (2019).
Liu, J. et al. TRIM28 is a distinct prognostic biomarker that worsens the tumor immune microenvironment in lung adenocarcinoma. Aging (Albany NY) 12, 20308–20331 (2020).
Zhang, Y. et al. SENP3 suppresses osteoclastogenesis by de-conjugating SUMO2/3 from IRF8 in bone marrow-derived monocytes. Cell. Rep. 30, 1951–1963.e1954 (2020).
Chang, T. H. et al. The small ubiquitin-like modifier-deconjugating enzyme sentrin-specific peptidase 1 switches IFN regulatory factor 8 from a repressor to an activator during macrophage activation. J. Immunol. 189, 3548–3556 (2012).
Masumi, A. & Ozato, K. Coactivator p300 acetylates the interferon regulatory factor-2 in U937 cells following phorbol ester treatment. J. Biol. Chem. 276, 20973–20980 (2001).
Qiu, W. et al. Sublytic C5b-9 triggers glomerular mesangial cell apoptosis via XAF1 gene activation mediated by p300-dependent IRF-1 acetylation. Cell. Death. Dis. 5, e1176 (2014).
Wu, Y. et al. Disrupting the phase separation of KAT8-IRF1 diminishes PD-L1 expression and promotes antitumor immunity. Nat. Cancer 4, 382–400 (2023).
Masumi, A. et al. Interferon regulatory factor-2 regulates cell growth through its acetylation. J. Biol. Chem. 278, 25401–25407 (2003).
Masumi, A. et al. Nucleolin is involved in interferon regulatory factor-2-dependent transcriptional activation. Oncogene 25, 5113–5124 (2006).
Wang, C. et al. The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J. Exp. Med. 214, 3597–3610 (2017).
Mino, T. & Takeuchi, O. NSD3 keeps IRF3 active. J. Exp. Med. 214, 3475–3476 (2017).
Wang, J. et al. Arginine methylation by PRMT2 promotes IFN-β production through TLR4/IRF3 signaling pathway. Mol. Immunol. 139, 202–210 (2021).
Huai, W. et al. KAT8 selectively inhibits antiviral immunity by acetylating IRF3. J. Exp. Med. 216, 772–785 (2019).
Caillaud, A. et al. Acetylation of interferon regulatory factor-7 by p300/CREB-binding protein (CBP)-associated factor (PCAF) impairs its DNA binding. J. Biol. Chem. 277, 49417–49421 (2002).
Qin, Z. et al. Deactylation by SIRT1 enables liquid-liquid phase separation of IRF3/IRF7 in innate antiviral immunity. Nat. Immunol. 23, 1193–1207 (2022).
Acidereli, H., Turut, F. A. & Cevik, O. Acetylation of interferon regulatory factor-5 suppresses androgen receptor and downregulates expression of Sox2. Cell. Biochem. Funct. 39, 667–678 (2021).
Tang, X. et al. Acetylation-dependent signal transduction for type I interferon receptor. Cell 131, 93–105 (2007).
Worbs, T., Hammerschmidt, S. I. & Förster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 17, 30–48 (2017).
Collin, M. & Bigley, V. Human dendritic cell subsets: an update. Immunology 154, 3–20 (2018).
Tailor, P., Tamura, T. & Ozato, K. IRF family proteins and type I interferon induction in dendritic cells. Cell. Res. 16, 134–140 (2006).
Suzuki, S. et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc. Natl Acad. Sci. USA 101, 8981–8986 (2004).
Tsujimura, H., Tamura, T. & Ozato, K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J. Immunol. 170, 1131–1135 (2003).
Tamura, T. et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J. Immunol. 174, 2573–2581 (2005).
Schiavoni, G. et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J. Exp. Med. 196, 1415–1425 (2002).
Aliberti, J. et al. Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood 101, 305–310 (2003).
Lança, T. et al. IRF8 deficiency induces the transcriptional, functional, and epigenetic reprogramming of cDC1 into the cDC2 lineage. Immunity 55, 1431–1447.e1411 (2022).
Gabriele, L. et al. IRF-1 deficiency skews the differentiation of dendritic cells toward plasmacytoid and tolerogenic features. J. Leukoc. Biol. 80, 1500–1511 (2006).
Ichikawa, E. et al. Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc. Natl Acad. Sci. USA 101, 3909–3914 (2004).
Negishi, H., Taniguchi, T. & Yanai, H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold. Spring. Harb. Perspect. Biol. 10, a028423 (2018).
Petro, T. M. IFN regulatory factor 3 in health and disease. J. Immunol. 205, 1981–1989 (2020).
Negishi, H. et al. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat. Immunol. 13, 659–666 (2012).
Matta, B., Song, S., Li, D. & Barnes, B. J. Interferon regulatory factor signaling in autoimmune disease. Cytokine 98, 15–26 (2017).
Qing, F. & Liu, Z. Interferon regulatory factor 7 in inflammation, cancer and infection. Front. Immunol. 14, 1190841 (2023).
Al Hamrashdi, M. & Brady, G. Regulation of IRF3 activation in human antiviral signaling pathways. Biochem. Pharmacol. 200, 115026 (2022).
Almuttaqi, H. & Udalova, I. A. Advances and challenges in targeting IRF5, a key regulator of inflammation. FEBS J. 286, 1624–1637 (2019).
Lazear, H. M. et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS. Pathog. 9, e1003118 (2013).
Panda, D. et al. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front. Immunol. 10, 1019 (2019).
Xia, X., Wang, W., Yin, K. & Wang, S. Interferon regulatory factor 8 governs myeloid cell development. Cytokine Growth Factor. Rev. 55, 48–57 (2020).
Tait Wojno, E. D., Hunter, C. A. & Stumhofer, J. S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 50, 851–870 (2019).
Verstockt, B. et al. IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 20, 433–446 (2023).
Kröger, A. et al. Activities of IRF-1. J. Interferon Cytokine Res. 22, 5–14 (2002).
Ng, L. G., Liu, Z., Kwok, I. & Ginhoux, F. Origin and heterogeneity of tissue myeloid cells: A focus on GMP-derived monocytes and neutrophils. Annu. Rev. Immunol. 41, 375–404 (2023).
Holtschke, T. et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 87, 307–317 (1996).
Scheller, M. et al. Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood 94, 3764–3771 (1999).
Schmidt, M. et al. The interferon regulatory factor ICSBP/IRF-8 in combination with PU.1 up-regulates expression of tumor suppressor p15(Ink4b) in murine myeloid cells. Blood 103, 4142–4149 (2004).
Diaz-Blanco, E. et al. Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia 21, 494–504 (2007).
Zhao, Y. et al. mTOR masters monocyte development in bone marrow by decreasing the inhibition of STAT5 on IRF8. Blood 131, 1587–1599 (2018).
Li, P. et al. IRF8 and IRF3 cooperatively regulate rapid interferon-β induction in human blood monocytes. Blood 117, 2847–2854 (2011).
Rosenbauer, F. et al. Disabled-2 is transcriptionally regulated by ICSBP and augments macrophage spreading and adhesion. EMBO J. 21, 211–220 (2002).
Tamura, T. et al. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood 106, 1938–1947 (2005).
Dror, N. et al. Identification of IRF-8 and IRF-1 target genes in activated macrophages. Mol. Immunol. 44, 338–346 (2007).
Tamura, T. et al. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 13, 155–165 (2000).
Karki, R. et al. IRF8 regulates transcription of naips for NLRC4 inflammasome activation. Cell 173, 920–933.e913 (2018).
Gupta, M. et al. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nat. Commun. 6, 6379 (2015).
Sjöstrand, M. et al. Expression of the immune regulator tripartite-motif 21 is controlled by IFN regulatory factors. J. Immunol. 191, 3753–3763 (2013).
Blanco, J. C. et al. Interferon regulatory factor (IRF)-1 and IRF-2 regulate interferon gamma-dependent cyclooxygenase 2 expression. J. Exp. Med. 191, 2131–2144 (2000).
Hobart, M. et al. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J. Immunol. 158, 4260–4269 (1997).
Kimura, T. et al. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science 264, 1921–1924 (1994).
Blériot, C., Chakarov, S. & Ginhoux, F. Determinants of resident tissue macrophage identity and function. Immunity 52, 957–970 (2020).
Park, M. D., Silvin, A., Ginhoux, F. & Merad, M. Macrophages in health and disease. Cell 185, 4259–4279 (2022).
Colonna, M. & Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468 (2017).
Borst, K., Dumas, A. A. & Prinz, M. Microglia: Immune and non-immune functions. Immunity 54, 2194–2208 (2021).
Zhou, N. et al. Transcriptional mechanism of IRF8 and PU.1 governs microglial activation in neurodegenerative condition. Protein Cell. 10, 87–103 (2019).
Masuda, T. et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell. Rep. 1, 334–340 (2012).
Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020).
Masuda, T. et al. Transcription factor IRF1 is responsible for IRF8-mediated IL-1β expression in reactive microglia. J. Pharmacol. Sci. 128, 216–220 (2015).
Feinberg, P. A. et al. Elevated TNF-α leads to neural circuit instability in the absence of interferon regulatory factor 8. J. Neurosci. 42, 6171–6185 (2022).
Ivashkiv, L. B. Metabolic-epigenetic coupling in osteoclast differentiation. Nat. Med. 21, 212–213 (2015).
Zhao, B. et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 15, 1066–1071 (2009).
Izawa, N. et al. Cooperation of PU.1 with IRF8 and NFATc1 defines chromatin landscapes during RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 34, 1143–1154 (2019).
Saito, E. et al. Down-regulation of Irf8 by Lyz2-cre/loxP accelerates osteoclast differentiation in vitro. Cytotechnology 69, 443–450 (2017).
Thumbigere-Math, V. et al. Inactivating mutation in IRF8 promotes osteoclast transcriptional programs and increases susceptibility to tooth root resorption. J. Bone Miner. Res. 34, 1155–1168 (2019).
Xia, Y. et al. TGFβ reprograms TNF stimulation of macrophages towards a non-canonical pathway driving inflammatory osteoclastogenesis. Nat. Commun. 13, 3920 (2022).
Gabrilovich, D. I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 5, 3–8 (2017).
Li, K. et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal. Transduct. Target. Ther. 6, 362 (2021).
Wu, Y. et al. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol. Cancer 21, 184 (2022).
Waight, J. D. et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J. Clin. Invest. 123, 4464–4478 (2013).
Paschall, A. V. et al. IFN regulatory factor 8 represses GM-CSF expression in T cells to affect myeloid cell lineage differentiation. J. Immunol. 194, 2369–2379 (2015).
Yang, J. et al. Cutting edge: IRF8 regulates Bax transcription in vivo in primary myeloid cells. J. Immunol. 187, 4426–4430 (2011).
Cumbo, C. et al. IRF4 expression is low in Philadelphia negative myeloproliferative neoplasms and is associated with a worse prognosis. Exp. Hematol. Oncol. 10, 58 (2021).
Lu, J., Liang, T., Li, P. & Yin, Q. Regulatory effects of IRF4 on immune cells in the tumor microenvironment. Front. Immunol. 14, 1086803 (2023).
Nam, S. et al. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J. Leukoc. Biol. 100, 1273–1284 (2016).
Yang, Q. et al. IRF7 regulates the development of granulocytic myeloid-derived suppressor cells through S100A9 transrepression in cancer. Oncogene 36, 2969–2980 (2017).
Lian, R. H. & Kumar, V. Murine natural killer cell progenitors and their requirements for development. Semin. Immunol. 14, 453–460 (2002).
Taki, S. et al. IFN regulatory factor-2 deficiency revealed a novel checkpoint critical for the generation of peripheral NK cells. J. Immunol. 174, 6005–6012 (2005).
Maffei, R. et al. The dynamic functions of IRF4 in B cell malignancies. Clin. Exp. Med. 23, 1171–1180 (2023).
Cook, S. L., Franke, M. C., Sievert, E. P. & Sciammas, R. A Synchronous IRF4-Dependent Gene Regulatory Network in B and Helper T Cells Orchestrating the Antibody Response. Trends Immunol. 41, 614–628 (2020).
Lu, R., Medina, K. L., Lancki, D. W. & Singh, H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes. Dev. 17, 1703–1708 (2003).
Cattoretti, G. et al. Stages of germinal center transit are defined by B cell transcription factor coexpression and relative abundance. J. Immunol. 177, 6930–6939 (2006).
Falini, B. et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood 95, 2084–2092 (2000).
Sciammas, R. et al. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25, 225–236 (2006).
Zhan, F. et al. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 101, 1128–1140 (2003).
Lee, C. H. et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J. Exp. Med. 203, 63–72 (2006).
Ashby, K. M. & Hogquist, K. A. A guide to thymic selection of T cells. Nat. Rev. Immunol. 24, 103–117 (2024).
Shrikant, P. A. et al. Regulating functional cell fates in CD8 T cells. Immunol. Res. 46, 12–22 (2010).
Visekruna, A. et al. Tc9 cells, a new subset of CD8(+) T cells, support Th2-mediated airway inflammation. Eur. J. Immunol. 43, 606–618 (2013).
Yen, H. R. et al. Tc17 CD8 T cells: functional plasticity and subset diversity. J. Immunol. 183, 7161–7168 (2009).
Huber, M. et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J. Clin. Invest. 123, 247–260 (2013).
Matsuyama, T. et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97 (1993).
Hida, S. et al. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity 13, 643–655 (2000).
Taki, S. et al. Multistage regulation of Th1-type immune responses by the transcription factor IRF-1. Immunity 6, 673–679 (1997).
Hida, S., Tadachi, M., Saito, T. & Taki, S. Negative control of basophil expansion by IRF-2 critical for the regulation of Th1/Th2 balance. Blood 106, 2011–2017 (2005).
Theofilopoulos, A. N., Koundouris, S., Kono, D. H. & Lawson, B. R. The role of IFN-gamma in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res. 3, 136–141 (2001).
Feng, D. et al. Irf5-deficient mice are protected from pristane-induced lupus via increased Th2 cytokines and altered IgG class switching. Eur. J. Immunol. 42, 1477–1487 (2012).
Brune, Z., Rice, M. R. & Barnes, B. J. Potential T cell-intrinsic regulatory roles for IRF5 via cytokine modulation in T helper subset differentiation and function. Front. Immunol. 11, 1143 (2020).
Walker, J. A. & McKenzie, A. N. J. T(H)2 cell development and function. Nat. Rev. Immunol. 18, 121–133 (2018).
Biswas, P. S., Bhagat, G. & Pernis, A. B. IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunol. Rev. 233, 79–96 (2010).
Honma, K. et al. Interferon regulatory factor 4 differentially regulates the production of Th2 cytokines in naive vs. effector/memory CD4+ T cells. Proc. Natl Acad. Sci. USA 105, 15890–15895 (2008).
Jefferies, C. A. Regulating IRFs in IFN driven disease. Front. Immunol. 10, 325 (2019).
Yagi, R., Zhu, J. & Paul, W. E. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int. Immunol. 23, 415–420 (2011).
Zhu, J. et al. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat. Immunol. 5, 1157–1165 (2004).
Fang, C. M. et al. Unique contribution of IRF-5-Ikaros axis to the B-cell IgG2a response. Genes. Immun. 13, 421–430 (2012).
Huber, M. & Lohoff, M. IRF4 at the crossroads of effector T-cell fate decision. Eur. J. Immunol. 44, 1886–1895 (2014).
Jabeen, R. et al. Th9 cell development requires a BATF-regulated transcriptional network. J. Clin. Invest. 123, 4641–4653 (2013).
Chang, H. C. et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 11, 527–534 (2010).
Brüstle, A. et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966 (2007).
Wang, J., Zhao, X. & Wan, Y. Y. Intricacies of TGF-β signaling in Treg and Th17 cell biology. Cell. Mol. Immunol. 20, 1002–1022 (2023).
Mudter, J. et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J. Clin. Invest. 118, 2415–2426 (2008).
Mudter, J. et al. IRF4 regulates IL-17A promoter activity and controls RORγt-dependent Th17 colitis in vivo. Inflamm. Bowel. Dis. 17, 1343–1358 (2011).
Huber, M. et al. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc. Natl. Acad. Sci. USA 105, 20846–20851 (2008).
Gutiérrez-Melo, N. & Baumjohann, D. T follicular helper cells in cancer. Trends Cancer 9, 309–325 (2023).
Liu, X., Nurieva, R. I. & Dong, C. Transcriptional regulation of follicular T-helper (Tfh) cells. Immunol. Rev. 252, 139–145 (2013).
Kwon, H. et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 31, 941–952 (2009).
Ise, W. et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 12, 536–543 (2011).
Betz, B. C. et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J. Exp. Med. 207, 933–942 (2010).
Ponnusamy, K. et al. The innate sensor ZBP1-IRF3 axis regulates cell proliferation in multiple myeloma. Haematologica 107, 721–732 (2022).
Hu, G. & Barnes, B. J. IRF-5 is a mediator of the death receptor-induced apoptotic signaling pathway. J. Biol. Chem. 284, 2767–2777 (2009).
Massimino, M. et al. IRF5 promotes the proliferation of human thyroid cancer cells. Mol. Cancer 11, 21 (2012).
Botti, E. et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc. Natl Acad. Sci. USA 108, 13710–13715 (2011).
Xu, L. et al. The developmental transcription factor IRF6 attenuates ABCG2 gene expression and distinctively reverses stemness phenotype in nasopharyngeal carcinoma. Cancer Lett. 431, 230–243 (2018).
Ma, X. et al. Inhibition of KIF20A by transcription factor IRF6 affects the progression of renal clear cell carcinoma. Cancer Cell. Int. 21, 246 (2021).
Lu, J. et al. Lin28A promotes IRF6-regulated aerobic glycolysis in glioma cells by stabilizing SNHG14. Cell. Death. Dis. 11, 447 (2020).
Chattopadhyay, S. & Sen, G. C. RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): a new antiviral pathway. Protein Cell. 8, 165–168 (2017).
Chattopadhyay, S., Yamashita, M., Zhang, Y. & Sen, G. C. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 85, 3708–3716 (2011).
White, C. L., Chattopadhyay, S. & Sen, G. C. Phosphatidylinositol 3-kinase signaling delays sendai virus-induced apoptosis by preventing XIAP degradation. J. Virol. 85, 5224–5227 (2011).
Sanz-Garcia, C. et al. The non-transcriptional activity of IRF3 modulates hepatic immune cell populations in acute-on-chronic ethanol administration in mice. J. Hepatol. 70, 974–984 (2019).
Wang, L. et al. Mechanisms of PANoptosis and relevant small-molecule compounds for fighting diseases. Cell. Death. Dis. 14, 851 (2023).
Malireddi, R. K. S., Kesavardhana, S. & Kanneganti, T. D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 9, 406 (2019).
Sharma, B. R., Karki, R., Rajesh, Y. & Kanneganti, T. D. Immune regulator IRF1 contributes to ZBP1-, AIM2-, RIPK1-, and NLRP12-PANoptosome activation and inflammatory cell death (PANoptosis). J. Biol. Chem. 299, 105141 (2023).
Kuriakose, T., Zheng, M., Neale, G. & Kanneganti, T. D. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J. Immunol. 200, 1489–1495 (2018).
Man, S. M. et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 16, 467–475 (2015).
Karki, R. et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 184, 149–168.e117 (2021).
Sundaram, B. et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell 186, 2783–2801.e2720 (2023).
Karki, R. et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI. Insight. 5, e136720 (2020).
Zhuang, Y. et al. Bile acid-induced IRF3 phosphorylation mediates cell death, inflammatory responses, and fibrosis in cholestasis-induced liver and kidney injury via regulation of ZBP1. Hepatology 79, 752–767 (2024).
Kondo, S. et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289 (2002).
Ingraham, C. R. et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335–1340 (2006).
Richardson, R. J. et al. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 38, 1329–1334 (2006).
Restivo, G. et al. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 30, 4571–4585 (2011).
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Li, D. & Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 6, 291 (2021).
Honda, K. et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl Acad. Sci. Usa. 101, 15416–15421 (2004).
Kawai, T. et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5, 1061–1068 (2004).
del Fresno, C. et al. Interferon-β production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38, 1176–1186 (2013).
Takaoka, A. et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249 (2005).
Barnes, B. J., Kellum, M. J., Field, A. E. & Pitha, P. M. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell. Biol. 22, 5721–5740 (2002).
Feng, D. et al. Differential requirement of histone acetylase and deacetylase activities for IRF5-mediated proinflammatory cytokine expression. J. Immunol. 185, 6003–6012 (2010).
Yang, C. et al. CXCL4 synergizes with TLR8 for TBK1-IRF5 activation, epigenomic remodeling and inflammatory response in human monocytes. Nat. Commun. 13, 3426 (2022).
Pradhan, P. et al. TRAF6-IRF5 kinetics, TRIF, and biophysical factors drive synergistic innate responses to particle-mediated MPLA-CpG co-presentation. Sci. Adv. 7, eabd4235 (2021).
Heinz, L. X. et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature 581, 316–322 (2020).
Zhang, H. et al. SLC15A4 controls endolysosomal TLR7-9 responses by recruiting the innate immune adaptor TASL. Cell. Rep. 42, 112916 (2023).
Briard, B. et al. Fungal ligands released by innate immune effectors promote inflammasome activation during Aspergillus fumigatus infection. Nat. Microbiol. 4, 316–327 (2019).
Tailor, P. et al. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 27, 228–239 (2007).
Zhao, J. et al. IRF-8/interferon (IFN) consensus sequence-binding protein is involved in Toll-like receptor (TLR) signaling and contributes to the cross-talk between TLR and IFN-gamma signaling pathways. J. Biol. Chem. 281, 10073–10080 (2006).
Shi, G. et al. IRF-8/miR-451a regulates M-MDSC differentiation via the AMPK/mTOR signal pathway during lupus development. Cell. Death. Discov. 7, 179 (2021).
Li, D. et al. IRF8 Impacts Self-Renewal of Hematopoietic Stem Cells by Regulating TLR9 Signaling Pathway of Innate Immune Cells. Adv. Sci. (Weinh.). 8, e2101031 (2021).
Goubau, D., Deddouche, S. & Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Rehwinkel, J. & Gack, M. U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551 (2020).
Yoneyama, M. et al. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 32, 48–53 (2015).
Sakaguchi, S. et al. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem. Biophys. Res. Commun. 306, 860–866 (2003).
Panne, D., McWhirter, S. M., Maniatis, T. & Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282, 22816–22822 (2007).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Yin, X. et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell. Rep. 34, 108628 (2021).
Ruffner, H., Reis, L. F., Näf, D. & Weissmann, C. Induction of type I interferon genes and interferon-inducible genes in embryonal stem cells devoid of interferon regulatory factor 1. Proc. Natl Acad. Sci. Usa. 90, 11503–11507 (1993).
Rosain, J. et al. Human IRF1 governs macrophagic IFN-γ immunity to mycobacteria. Cell 186, 621–645.e633 (2023).
Ishii, K. J. et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48 (2006).
Takaoka, A. et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007).
Lei, Y. et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell 186, 3013–3032.e3022 (2023).
Di Paolo, N. C. et al. The transcription factor IRF3 triggers “defensive suicide” necrosis in response to viral and bacterial pathogens. Cell. Rep. 3, 1840–1846 (2013).
Cevik, O. et al. Interferon regulatory factor 5 (IRF5) suppresses hepatitis C virus (HCV) replication and HCV-associated hepatocellular carcinoma. J. Biol. Chem. 292, 21676–21689 (2017).
Nie, S. et al. The protective effect of interfering TLR9-IRF5 signaling pathway on the development of CVB3-induced myocarditis. Clin. Immunol. 207, 24–35 (2019).
Carmona-Pérez, L. et al. The TLR7/IRF-5 axis sensitizes memory CD4+ T cells to Fas-mediated apoptosis during HIV-1 infection. JCI. Insight. 8, e167329 (2023).
Liu, Y. et al. Airway acidification impaired host defense against Pseudomonas aeruginosa infection by promoting type 1 interferon β response. Emerg. Microbes Infect. 11, 2132–2146 (2022).
Puthia, M. et al. IRF7 inhibition prevents destructive innate immunity-A target for nonantibiotic therapy of bacterial infections. Sci. Transl. Med. 8, 336ra359 (2016).
Schoggins, J. W. et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014).
Mboko, W. P. et al. Interferon Regulatory Factor 1 and Type I Interferon Cooperate To Control Acute Gammaherpesvirus Infection. J. Virol. 91, e01444-16 (2017).
Feng, H., Zhang, Y. B. & Gui, J. F. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLoS. Pathog. 17, e1009220 (2021).
Loevenich, S. et al. Human Metapneumovirus Induces IRF1 via TANK-Binding Kinase 1 and Type I IFN. Front. Immunol. 12, 563336 (2021).
Yamane, D. et al. Basal expression of interferon regulatory factor 1 drives intrinsic hepatocyte resistance to multiple RNA viruses. Nat. Microbiol. 4, 1096–1104 (2019).
Wang, J. et al. IRF1 Promotes the Innate Immune Response to Viral Infection by Enhancing the Activation of IRF3. J. Virol. 94, e01231-20 (2020).
Ren, K. et al. IRF2 inhibits ZIKV replication by promoting FAM111A expression to enhance the host restriction effect of RFC3. Virol. J. 18, 256 (2021).
Panda, D. et al. Triad of human cellular proteins, IRF2, FAM111A, and RFC3, restrict replication of orthopoxvirus SPI-1 host-range mutants. Proc. Natl Acad. Sci. USA 114, 3720–3725 (2017).
Persyn, E. et al. IRF2 is required for development and functional maturation of human NK cells. Front. Immunol. 13, 1038821 (2022).
Glanz, A. et al. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses 13, 575 (2021).
Popli, S., Chakravarty, S., Fan, S. & Glanz, A. IRF3 inhibits nuclear translocation of NF-κB to prevent viral inflammation. Proc. Natl Acad. Sci. Usa. 119, e2121385119 (2022).
Canivet, C. et al. Both IRF3 and especially IRF7 play a key role to orchestrate an effective cerebral inflammatory response in a mouse model of herpes simplex virus encephalitis. J. Neurovirol. 24, 761–768 (2018).
Andersen, L. L. et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 212, 1371–1379 (2015).
Campbell, T. M. & Liu, Z. Respiratory viral infections in otherwise healthy humans with inherited IRF7 deficiency. J. Exp. Med. 219, e20220202 (2022).
Tucker, M. H. et al. IRF7 and UNC93B1 variants in an infant with recurrent herpes simplex virus infection. J. Clin. Invest. 133, e154016 (2023).
Clohisey, S. & Baillie, J. K. Host susceptibility to severe influenza A virus infection. Crit. Care. 23, 303 (2019).
Hernandez, N. & Melki, I. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 215, 2567–2585 (2018).
Ji, W. et al. TBK1 and IRF3 are potential therapeutic targets in Enterovirus A71-associated diseases. PLoS. Negl. Trop. Dis. 17, e0011001 (2023).
Man, K. et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141.e1125 (2017).
Grusdat, M. et al. IRF4 and BATF are critical for CD8+ T-cell function following infection with LCMV. Cell. Death. Differ. 21, 1050–1060 (2014).
Yanai, H. et al. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl Acad. Sci. USA 104, 3402–3407 (2007).
Thackray, L. B. et al. Interferon regulatory factor 5-dependent immune responses in the draining lymph node protect against West Nile virus infection. J. Virol. 88, 11007–11021 (2014).
Song, J. et al. Human cytomegalovirus induces and exploits Roquin to counteract the IRF1-mediated antiviral state. Proc. Natl Acad. Sci. USA 116, 18619–18628 (2019).
Ueki, I. F. et al. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J. Exp. Med. 210, 1929–1936 (2013).
Cheng, X. & Ratner, L. HIV-2 Vpx protein interacts with interferon regulatory factor 5 (IRF5) and inhibits its function. J. Biol. Chem. 289, 9146–9157 (2014).
Su, S. et al. Modulation of innate immune response to viruses including SARS-CoV-2 by progesterone. Signal. Transduct. Target. Ther. 7, 137 (2022).
Yoo, J. S. & Sasaki, M. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 12, 6602 (2021).
Hall, R. et al. SARS-CoV-2 ORF6 disrupts innate immune signalling by inhibiting cellular mRNA export. PLoS. Pathog. 18, e1010349 (2022).
Rashid, F. et al. Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front. Immunol. 13, 940756 (2022).
Moustaqil, M. & Ollivier, E. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg. Microbes Infect. 10, 178–195 (2021).
Shin, D. & Mukherjee, R. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Wang, W. et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell. Mol. Immunol. 18, 945–953 (2021).
Zheng, Y. et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal. Transduct. Target. Ther. 7, 22 (2022).
Feng, K. et al. SARS-CoV-2 NSP13 interacts with host IRF3, blocking antiviral immune responses. J. Med. Virol. 95, e28881 (2023).
Fung, S. Y. et al. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int. J. Biol. Sci. 17, 1547–1554 (2021).
Shin, J. et al. SARS-CoV-2 infection impairs the insulin/IGF signaling pathway in the lung, liver, adipose tissue, and pancreatic cells via IRF1. Metabolism 133, 155236 (2022).
Huang, H. C. et al. Upregulation of PD-L1 by SARS-CoV-2 promotes immune evasion. J. Med. Virol. 95, e28478 (2023).
Zhang, Q. & Bastard, P. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Lévy, R. & Zhang, P. Monoclonal antibody-mediated neutralization of SARS-CoV-2 in an IRF9-deficient child. Proc. Natl. Acad. Sci. USA. 118, e2114390118 (2021).
Mishra, R. & Banerjea, A. C. SARS-CoV-2 Spike Targets USP33-IRF9 Axis via Exosomal miR-148a to Activate Human Microglia. Front. Immunol. 12, 656700 (2021).
Gidon, A. et al. The Tumor Necrosis Factor Alpha and Interleukin 6 Auto-paracrine Signaling Loop Controls Mycobacterium avium Infection via Induction of IRF1/IRG1 in Human Primary Macrophages. mBio 12, e0212121 (2021).
Zhou, X. et al. Interferon regulatory factor 1 eliminates mycobacteria by suppressing p70 S6 kinase via mechanistic target of rapamycin signaling. J. Infect. 79, 262–276 (2019).
Schmalzl, A., Leupold, T. & Kreiss, L. Interferon regulatory factor 1 (IRF-1) promotes intestinal group 3 innate lymphoid responses during Citrobacter rodentium infection. Nat. Commun. 13, 5730 (2022).
Yoon, G. S. et al. Interferon regulatory factor-1 in flagellin-induced reprogramming: potential protective role of CXCL10 in cornea innate defense against Pseudomonas aeruginosa infection. Invest. Ophthalmol. Vis. Sci. 54, 7510–7521 (2013).
Li, Q., Liu, C., Yue, R. & El-Ashram, S. cGAS/STING/TBK1/IRF3 Signaling Pathway Activates BMDCs Maturation Following Mycobacterium bovis Infection. Int. J. Mol. Sci. 20, 895 (2019).
Cui, Y. et al. Mycobacterium bovis Induces Endoplasmic Reticulum Stress Mediated-Apoptosis by Activating IRF3 in a Murine Macrophage Cell Line. Front. Cell. Infect. Microbiol. 6, 182 (2016).
Cheng, Y. & Schorey, J. S. Mycobacterium tuberculosis-induced IFN-β production requires cytosolic DNA and RNA sensing pathways. J. Exp. Med. 215, 2919–2935 (2018).
Pandey, A. K. et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS. Pathog. 5, e1000500 (2009).
Zhang, Z. et al. MicroRNA-31 mediated by interferon regulatory factor 7 signaling facilitates control of Mycobacterium tuberculosis infection. Int. J. Med. Microbiol. 312, 151569 (2022).
Skjesol, A. & Yurchenko, M. The TLR4 adaptor TRAM controls the phagocytosis of Gram-negative bacteria by interacting with the Rab11-family interacting protein 2. PLoS. Pathog. 15, e1007684 (2019).
Dooyema, S. D. R. et al. Helicobacter pylori actively suppresses innate immune nucleic acid receptors. Gut. Microbes 14, 2105102 (2022).
Yu, X. et al. Cross-Regulation of Two Type I Interferon Signaling Pathways in Plasmacytoid Dendritic Cells Controls Anti-malaria Immunity and Host Mortality. Immunity 45, 1093–1107 (2016).
Yu, X. et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat. Commun. 9, 4964 (2018).
Swanson, R. V. et al. Antigen-specific B cells direct T follicular-like helper cells into lymphoid follicles to mediate Mycobacterium tuberculosis control. Nat. Immunol. 24, 855–868 (2023).
Harberts, A. & Schmidt, C. Interferon regulatory factor 4 controls effector functions of CD8(+) memory T cells. Proc. Natl. Acad. Sci. USA 118, e2014553118 (2021).
Pandey, S. P., Yan, J., Turner, J. R. & Abraham, C. Reducing IRF5 expression attenuates colitis in mice, but impairs the clearance of intestinal pathogens. Mucosal. Immunol. 12, 874–887 (2019).
Hedl, M., Yan, J. & Witt, H. IRF5 Is Required for Bacterial Clearance in Human M1-Polarized Macrophages, and IRF5 Immune-Mediated Disease Risk Variants Modulate This Outcome. J. Immunol. 202, 920–930 (2019).
Corbin, A. L., Gomez-Vazquez, M. & Berthold, D. L. IRF5 guides monocytes toward an inflammatory CD11c(+) macrophage phenotype and promotes intestinal inflammation. Sci. Immunol. 5, eaax6085 (2020).
Mathy, N. W. et al. The Long Non-Coding RNA Nostrill Regulates Transcription of Irf7 Through Interaction With NF-κB p65 to Enhance Intestinal Epithelial Defense Against Cryptosporidium parvum. Front. Immunol. 13, 863957 (2022).
Karki, R., Lee, E. & Sharma, B. R. IRF8 Regulates Gram-Negative Bacteria-Mediated NLRP3 Inflammasome Activation and Cell Death. J. Immunol. 204, 2514–2522 (2020).
Biondo, C. et al. Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur. J. Immunol. 42, 2632–2643 (2012).
Yang, L. et al. InsP(3)R-SEC5 interaction on phagosomes modulates innate immunity to Candida albicans by promoting cytosolic Ca(2+) elevation and TBK1 activity. Bmc. Biol. 16, 46 (2018).
Brown Harding, H. et al. Candida albicans extracellular vesicles trigger type I IFN signalling via cGAS and STING. Nat. Microbiol. 9, 95–107 (2024).
Li, F. et al. C-type lectin receptor 2d forms homodimers and heterodimers with TLR2 to negatively regulate IRF5-mediated antifungal immunity. Nat. Commun. 14, 6718 (2023).
Qing, F. et al. IRF7 Exacerbates Candida albicans Infection by Compromising CD209-Mediated Phagocytosis and Autophagy-Mediated Killing in Macrophages. J. Immunol. 212, 1932–1944 (2024).
Wevers, B. A. et al. Fungal engagement of the C-type lectin mincle suppresses dectin-1-induced antifungal immunity. Cell. Host. Microbe 15, 494–505 (2014).
Wiesner, D. L. et al. Regulatory T Cell Induction and Retention in the Lungs Drives Suppression of Detrimental Type 2 Th Cells During Pulmonary Cryptococcal Infection. J. Immunol. 196, 365–374 (2016).
Valdez, P. A. et al. Prostaglandin E2 suppresses antifungal immunity by inhibiting interferon regulatory factor 4 function and interleukin-17 expression in T cells. Immunity 36, 668–679 (2012).
Harada, H. et al. Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science 259, 971–974 (1993).
van der Weyden, L. et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 541, 233–236 (2017).
Jeong, S. I. et al. XAF1 forms a positive feedback loop with IRF-1 to drive apoptotic stress response and suppress tumorigenesis. Cell. Death. Dis. 9, 806 (2018).
Yanai, H., Negishi, H. & Taniguchi, T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology 1, 1376–1386 (2012).
Shao, L., Hou, W. & Scharping, N. E. IRF1 Inhibits Antitumor Immunity through the Upregulation of PD-L1 in the Tumor Cell. Cancer Immunol. Res. 7, 1258–1266 (2019).
Kriegsman, B. A. & Vangala, P. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J. Immunol. 203, 1999–2010 (2019).
Arnold, M. et al. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 159, 335–349.e315 (2020).
Wang, Y. et al. Involvement of IFN regulatory factor (IRF)-1 and IRF-2 in the formation and progression of human esophageal cancers. Cancer Res. 67, 2535–2543 (2007).
Wang, Y. et al. Negative feedback regulation of IFN-gamma pathway by IFN regulatory factor 2 in esophageal cancers. Cancer Res. 68, 1136–1143 (2008).
Huang, J. et al. Interferon-inducible lncRNA IRF1-AS represses esophageal squamous cell carcinoma by promoting interferon response. Cancer Lett. 459, 86–99 (2019).
Watson, G. A. et al. Ad-IRF-1 induces apoptosis in esophageal adenocarcinoma. Neoplasia 8, 31–37 (2006).
Zhou, Y. et al. FOXM1c promotes oesophageal cancer metastasis by transcriptionally regulating IRF1 expression. Cell. Prolif. 52, e12553 (2019).
Wierstra, I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv. Cancer Res. 119, 191–419 (2013).
Zhang, C. F. et al. Relationship between polymorphism of IRF-3 gene codon 427 and esophageal cancer in Anyang population of China. Beijing. Da. Xue. Xue. Bao. Yi. Xue. Ban. 36, 345–347 (2004).
Dai, W. et al. Whole-exome sequencing reveals critical genes underlying metastasis in oesophageal squamous cell carcinoma. J. Pathol. 242, 500–510 (2017).
Chen, J. et al. Genomic profiling of 766 cancer-related genes in archived esophageal normal and carcinoma tissues. Int. J. Cancer 122, 2249–2254 (2008).
Sun, R. et al. Prognostic significance of interferon regulating factor 4 in esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 506, 685–691 (2018).
Nozawa, H. et al. Functionally inactivating point mutation in the tumor-suppressor IRF-1 gene identified in human gastric cancer. Int. J. Cancer 77, 522–527 (1998).
Gao, J. et al. IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell. Death. Differ. 17, 699–709 (2010).
Gao, J., Tian, Y. & Zhang, J. Overexpression of interferon regulatory factor 1 enhances chemosensitivity to 5-fluorouracil in gastric cancer cells. J. Cancer Res. Ther. 8, 57–61 (2012).
Yuan, J. et al. Interferon regulatory factor-1 reverses chemoresistance by downregulating the expression of P-glycoprotein in gastric cancer. Cancer Lett. 457, 28–39 (2019).
Tan, L. et al. Interferon regulatory factor-1 suppresses DNA damage response and reverses chemotherapy resistance by downregulating the expression of RAD51 in gastric cancer. Am. J. Cancer Res. 10, 1255–1270 (2020).
Yuan, J. et al. MIR17HG-miR-18a/19a axis, regulated by interferon regulatory factor-1, promotes gastric cancer metastasis via Wnt/β-catenin signalling. Cell. Death. Dis. 10, 454 (2019).
Chen, Y. J. et al. MicroRNA-18a modulates P53 expression by targeting IRF2 in gastric cancer patients. J. Gastroenterol. Hepatol. 31, 155–163 (2016).
Chen, Y. J. et al. IRF-2 Inhibits Gastric Cancer Invasion and Migration by Down-Regulating MMP-1. Dig. Dis. Sci. 65, 168–177 (2020).
Chen, Y. J. et al. IRF-2 inhibits cancer proliferation by promoting AMER-1 transcription in human gastric cancer. J. Transl. Med. 20, 68 (2022).
Jiao, S. et al. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J. Exp. Med. 215, 699–718 (2018).
Qiao, Y., Li, T., Zheng, S. & Wang, H. The Hippo pathway as a drug target in gastric cancer. Cancer Lett. 420, 14–25 (2018).
Zhang, T. et al. microRNA-365 inhibits YAP through TLR4-mediated IRF3 phosphorylation and thereby alleviates gastric precancerous lesions. Cancer Cell. Int. 20, 549 (2020).
Yamashita, M. et al. DNA methylation of interferon regulatory factors in gastric cancer and noncancerous gastric mucosae. Cancer Sci. 101, 1708–1716 (2010).
Jee, C. D. et al. Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur. J. Cancer 45, 1282–1293 (2009).
Li, D. et al. IRF6 Is Directly Regulated by ZEB1 and ELF3, and Predicts a Favorable Prognosis in Gastric Cancer. Front. Oncol. 9, 220 (2019).
Du, J. et al. Cytoplasmic localization of IRF5 induces Wnt5a/E-cadherin degradation and promotes gastric cancer cells metastasis. Cancer Gene. Ther. 30, 866–877 (2023).
Xing, Y., Chen, H., Guo, Z. & Zhou, X. Circular RNA circ0007360 Attenuates Gastric Cancer Progression by Altering the miR-762/IRF7 Axis. Front. Cell. Dev. Biol. 10, 789073 (2022).
Fu, K. et al. Single-cell RNA sequencing of immune cells in gastric cancer patients. Aging (Albany NY) 12, 2747–2763 (2020).
Yan, Y. et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 regulate PD-L1 expression in hepatocellular carcinoma (HCC) cells. Cancer Immunol. Immunother. 69, 1891–1903 (2020).
Lin, Y. H. et al. Repression of microRNA-130b by thyroid hormone enhances cell motility. J. Hepatol. 62, 1328–1340 (2015).
Wang, R. et al. Interferon Gamma-Induced Interferon Regulatory Factor 1 Activates Transcription of HHLA2 and Induces Immune Escape of Hepatocellular Carcinoma Cells. Inflammation 45, 308–330 (2022).
Wang, Z. & Pan, B. SUMOylated IL-33 in the nucleus stabilizes the transcription factor IRF1 in hepatocellular carcinoma cells to promote immune escape. Sci. Signal. 16, eabq3362 (2023).
Yu, M. et al. miR-345 inhibits tumor metastasis and EMT by targeting IRF1-mediated mTOR/STAT3/AKT pathway in hepatocellular carcinoma. Int. J. Oncol. 50, 975–983 (2017).
Li, P. et al. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 314, 213–222 (2012).
Kröger, A. et al. Growth suppression of the hepatocellular carcinoma cell line Hepa1-6 by an activatable interferon regulatory factor-1 in mice. Cancer Res. 61, 2609–2617 (2001).
Zekri, A. R. et al. Disease progression from chronic hepatitis C to cirrhosis and hepatocellular carcinoma is associated with repression of interferon regulatory factor-1. Eur. J. Gastroenterol. Hepatol. 22, 450–456 (2010).
Yi, Y. et al. Interferon regulatory factor (IRF)-1 and IRF-2 are associated with prognosis and tumor invasion in HCC. Ann. Surg. Oncol. 20, 267–276 (2013).
Moriyama, Y. et al. Tumor-suppressor effect of interferon regulatory factor-1 in human hepatocellular carcinoma. Clin. Cancer Res. 7, 1293–1298 (2001).
Yan, Y. et al. Interferon regulatory factor 1 (IRF-1) downregulates Checkpoint kinase 1 (CHK1) through miR-195 to upregulate apoptosis and PD-L1 expression in Hepatocellular carcinoma (HCC) cells. Br. J. Cancer 125, 101–111 (2021).
Yan, Y. et al. Interferon regulatory factor 1(IRF-1) activates anti-tumor immunity via CXCL10/CXCR3 axis in hepatocellular carcinoma (HCC). Cancer Lett. 506, 95–106 (2021).
Wan, P. Q. et al. Analysis of the relationship between microRNA-31 and interferon regulatory factor-1 in hepatocellular carcinoma cells. Eur. Rev. Med. Pharmacol. Sci. 24, 647–654 (2020).
Yan, Y. et al. MicroRNA-23a downregulates the expression of interferon regulatory factor-1 in hepatocellular carcinoma cells. Oncol. Rep. 36, 633–640 (2016).
Dong, K. et al. MicroRNA-301a (miR-301a) is induced in hepatocellular carcinoma (HCC) and down- regulates the expression of interferon regulatory factor-1. Biochem. Biophys. Res. Commun. 524, 273–279 (2020).
Yu, W. et al. NR4A1 mediates NK-cell dysfunction in hepatocellular carcinoma via the IFN-γ/p-STAT1/IRF1 pathway. Immunology 169, 69–82 (2023).
Guichard, C. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 44, 694–698 (2012).
Guo, Y. et al. IRF2 regulates cellular survival and Lenvatinib-sensitivity of hepatocellular carcinoma (HCC) through regulating β-catenin. Transl. Oncol. 14, 101059 (2021).
Yu, H. et al. Major Vault Protein Promotes Hepatocellular Carcinoma Through Targeting Interferon Regulatory Factor 2 and Decreasing p53 Activity. Hepatology 72, 518–534 (2020).
Yuan, M. M. et al. TLR3 expression correlates with apoptosis, proliferation and angiogenesis in hepatocellular carcinoma and predicts prognosis. Bmc. Cancer 15, 245 (2015).
Qi, Z. et al. Identification of prognostic biomarkers and correlations with immune infiltrates among cGAS-STING in hepatocellular carcinoma. Biosci. Rep. 40, BSR20202603 (2020).
Kim, G. W. & Imam, H. HBV-Induced Increased N6 Methyladenosine Modification of PTEN RNA Affects Innate Immunity and Contributes to HCC. Hepatology 73, 533–547 (2021).
Du, S. S. et al. Radiation Therapy Promotes Hepatocellular Carcinoma Immune Cloaking via PD-L1 Upregulation Induced by cGAS-STING Activation. Int. J. Radiat. Oncol. Biol. Phys. 112, 1243–1255 (2022).
Ma, H., Kang, Z. & Foo, T. K. Disrupted BRCA1-PALB2 interaction induces tumor immunosuppression and T-lymphocyte infiltration in HCC through cGAS-STING pathway. Hepatology 77, 33–47 (2023).
Yuan, J. et al. Construction and validation of an IRF4 risk score to predict prognosis and response to immunotherapy in hepatocellular carcinoma. Int. Immunopharmacol. 113, 109411 (2022).
Shin, S. H. et al. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array. J. Korean Med. Sci. 25, 1152–1159 (2010).
Yu, J. et al. Methylation profiling of twenty four genes and the concordant methylation behaviours of nineteen genes that may contribute to hepatocellular carcinogenesis. Cell. Res. 13, 319–333 (2003).
Wu, H. et al. Hepatic interferon regulatory factor 8 expression suppresses hepatocellular carcinoma progression and enhances the response to anti-programmed cell death protein-1 therapy. Hepatology 76, 1602–1616 (2022).
Tamada, Y. et al. p48 Overexpression enhances interferon-mediated expression and activity of double-stranded RNA-dependent protein kinase in human hepatoma cells. J. Hepatol. 37, 493–499 (2002).
Wu, W. Z. et al. Reduction in p48-ISGFgamma levels confers resistance to interferon-alpha2a in MHCC97 cells. Oncology 67, 428–440 (2004).
Qian, Y. B. et al. P48 is a predictive marker for outcome of postoperative interferon-alpha treatment in patients with hepatitis B virus infection-related hepatocellular carcinoma. Cancer 107, 1562–1569 (2006).
Huang, X., Zhang, G. & Liang, T. Subtyping for pancreatic cancer precision therapy. Trends Pharmacol. Sci. 43, 482–494 (2022).
Sakai, T. et al. The roles of interferon regulatory factors 1 and 2 in the progression of human pancreatic cancer. Pancreas 43, 909–916 (2014).
Hannes, S., Karlowitz, R. & van Wijk, S. J. L. The Smac mimetic BV6 cooperates with STING to induce necroptosis in apoptosis-resistant pancreatic carcinoma cells. Cell. Death. Dis. 12, 816 (2021).
Somerville, T. D. D., Xu, Y., Wu, X. S. & Maia-Silva, D. ZBED2 is an antagonist of interferon regulatory factor 1 and modifies cell identity in pancreatic cancer. Proc. Natl Acad. Sci. Usa. 117, 11471–11482 (2020).
Cui, L. et al. IRF-2 is over-expressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 33, 247–255 (2012).
Zhang, K. et al. Comprehensive analysis of expression profile and prognostic significance of interferon regulatory factors in pancreatic cancer. Bmc. Genom. Data. 23, 5 (2022).
Kou, Y. Q. et al. Prognostic-Related Biomarkers in Pancreatic Ductal Adenocarcinoma Correlating with Immune Infiltrates Based on Proteomics. Med. Sci. Monit. 29, e938785 (2023).
Liu, W. et al. Identifying a novel IRF3/circUHRF1/miR-1306-5p/ARL4C axis in pancreatic ductal adenocarcinoma progression. Cell. Cycle 21, 392–405 (2022).
Xie, W. & Li, X. The Pyroptosis-Related Gene Prognostic Index Associated with Tumor Immune Infiltration for Pancreatic Cancer. Int. J. Mol. Sci. 23, 6178 (2022).
Metzger, P. & Kirchleitner, S. V. Systemic but not MDSC-specific IRF4 deficiency promotes an immunosuppressed tumor microenvironment in a murine pancreatic cancer model. Cancer Immunol. Immunother. 69, 2101–2112 (2020).
Muthalagu, N. & Monteverde, T. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 10, 872–887 (2020).
Meyer, M. A. et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat. Commun. 9, 1250 (2018).
Hong, M. et al. IRF1 inhibits the proliferation and metastasis of colorectal cancer by suppressing the RAS-RAC1 pathway. Cancer Manag. Res. 11, 369–378 (2019).
Wu, Y., Zhang, S. & Yan, J. IRF1 association with tumor immune microenvironment and use as a diagnostic biomarker for colorectal cancer recurrence. Oncol. Lett. 19, 1759–1770 (2020).
Yuan, L. et al. IRF1 Inhibits Autophagy-Mediated Proliferation of Colorectal Cancer via Targeting ATG13. Cancer Invest. 40, 35–45 (2022).
Sun, B. et al. Colorectal cancer exosomes induce lymphatic network remodeling in lymph nodes. Int. J. Cancer 145, 1648–1659 (2019).
Liao, W. et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 35, 559–572.e557 (2019).
Chen, Y. J. et al. Interferon regulatory factor family influences tumor immunity and prognosis of patients with colorectal cancer. J. Transl. Med. 19, 379 (2021).
You, W., Di, A., Zhang, L. & Zhao, G. Effects of wogonin on the growth and metastasis of colon cancer through the Hippo signaling pathway. Bioengineered 13, 2586–2597 (2022).
Tian, M. et al. IRF3 prevents colorectal tumorigenesis via inhibiting the nuclear translocation of β-catenin. Nat. Commun. 11, 5762 (2020).
Ding, C. et al. β-catenin regulates IRF3-mediated innate immune signalling in colorectal cancer. Cell. Prolif. 51, e12464 (2018).
Corrigendum. Cell Prolif 54, e12655, (2021).
Kießler, M. et al. Tumor-infiltrating plasmacytoid dendritic cells are associated with survival in human colon cancer. J. Immunother. Cancer. 9, e001813 (2021).
Roulois, D. et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162, 961–973 (2015).
Udden, S. M. N. et al. NOD2 Suppresses Colorectal Tumorigenesis via Downregulation of the TLR Pathways. Cell. Rep. 19, 2756–2770 (2017).
Zhou, Y. et al. Integrated multi-omics data analyses for exploring the co-occurring and mutually exclusive gene alteration events in colorectal cancer. Hum. Mutat. 41, 1588–1599 (2020).
Hu, G. & Barnes, B. J. Interferon regulatory factor-5-regulated pathways as a target for colorectal cancer therapeutics. Expert. Rev. Anticancer. Ther. 6, 775–784 (2006).
Arnold, I. C. et al. The GM-CSF-IRF5 signaling axis in eosinophils promotes antitumor immunity through activation of type 1 T cell responses. J. Exp. Med. 217, e20190706 (2020).
Tan, L. et al. The interferon regulatory factor 6 promotes cisplatin sensitivity in colorectal cancer. Bioengineered 13, 10504–10517 (2022).
Ibrahim, M. L. et al. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell. Rep. 25, 3036–3046.e3036 (2018).
Klement, J. D. et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest. 128, 5549–5560 (2018).
Sharma, B. R. et al. The Transcription Factor IRF9 Promotes Colorectal Cancer via Modulating the IL-6/STAT3 Signaling Axis. Cancers (Basel) 14, 919 (2022).
Thai, A. A. et al. Lung cancer. Lancet 398, 535–554 (2021).
Lahiri, A. et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol. Cancer 22, 40 (2023).
Huang, J. X. et al. IRF1 Negatively Regulates Oncogenic KPNA2 Expression Under Growth Stimulation and Hypoxia in Lung Cancer Cells. Onco. Targets Ther. 12, 11475–11486 (2019).
Zhang, L. et al. Interferon regulatory factor-1 regulates cisplatin-induced apoptosis and autophagy in A549 lung cancer cells. Med. Oncol. 39, 38 (2022).
Chan, Y. C. & Chang, Y. C. Overexpression of PSAT1 promotes metastasis of lung adenocarcinoma by suppressing the IRF1-IFNγ axis. Oncogene 39, 2509–2522 (2020).
Qi, L. et al. An individualized gene expression signature for prediction of lung adenocarcinoma metastases. Mol. Oncol. 11, 1630–1645 (2017).
Liang, C. et al. MicroRNA-18a-5p functions as an oncogene by directly targeting IRF2 in lung cancer. Cell. Death. Dis. 8, e2764 (2017).
Jin, J. J. et al. Overexpression of miR-1290 contributes to cell proliferation and invasion of non small cell lung cancer by targeting interferon regulatory factor 2. Int. J. Biochem. Cell. Biol. 95, 113–120 (2018).
Ma, J. et al. LncRNA GAS5 modulates the progression of non-small cell lung cancer through repressing miR-221-3p and up-regulating IRF2. Diagn. Pathol. 16, 46 (2021).
Xiao, X. H. & He, S. Y. ELF1 activated long non-coding RNA CASC2 inhibits cisplatin resistance of non-small cell lung cancer via the miR-18a/IRF-2 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 24, 3130–3142 (2020).
Liu, F. et al. Upregulation of microRNA-450 inhibits the progression of lung cancer in vitro and in vivo by targeting interferon regulatory factor 2. Int. J. Mol. Med. 38, 283–290 (2016).
Yi, L. et al. Interferon regulatory factor 3 mediates Poly(I:C)-induced innate immune response and apoptosis in non‑small cell lung cancer. Int. J. Oncol. 52, 1623–1632 (2018).
Gong, K. et al. EGFR inhibition triggers an adaptive response by co-opting antiviral signaling pathways in lung cancer. Nat. Cancer 1, 394–409 (2020).
Zhang, N. et al. PARP inhibitor niraparib as a radiosensitizer promotes antitumor immunity of radiotherapy in EGFR-mutated non-small cell lung cancer. Clin. Transl. Oncol. 23, 1827–1837 (2021).
Taniguchi, H. et al. WEE1 inhibition enhances the antitumor immune response to PD-L1 blockade by the concomitant activation of STING and STAT1 pathways in SCLC. Cell. Rep. 39, 110814 (2022).
Zhou, L. et al. Low-dose carboplatin reprograms tumor immune microenvironment through STING signaling pathway and synergizes with PD-1 inhibitors in lung cancer. Cancer Lett. 500, 163–171 (2021).
Sen, T. et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 9, 646–661 (2019).
Long, Y. et al. GPR162 activates STING dependent DNA damage pathway as a novel tumor suppressor and radiation sensitizer. Signal. Transduct. Target. Ther. 8, 48 (2023).
Wu, Y. Y. et al. CPEB4 and IRF4 expression in peripheral mononuclear cells are potential prognostic factors for advanced lung cancer. J. Formos. Med. Assoc. 116, 114–122 (2017).
Li, X. et al. Interferon Regulatory Factor 4 Correlated With Immune Cells Infiltration Could Predict Prognosis for Patients With Lung Adenocarcinoma. Front. Oncol. 11, 698465 (2021).
Chen, H. Y. et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N. Engl. J. Med. 356, 11–20 (2007).
Gao, J. et al. Exosomal circZNF451 restrains anti-PD1 treatment in lung adenocarcinoma via polarizing macrophages by complexing with TRIM56 and FXR1. J. Exp. Clin. Cancer Res. 41, 295 (2022).
Carbó, J. M. & León, T. E. Pharmacologic Activation of LXR Alters the Expression Profile of Tumor-Associated Macrophages and the Abundance of Regulatory T Cells in the Tumor Microenvironment. Cancer Res. 81, 968–985 (2021).
Alvisi, G. et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Invest. 130, 3137–3150 (2020).
Feng, D. D. et al. Transcription factor E2F1 positively regulates interferon regulatory factor 5 expression in non-small cell lung cancer. Onco. Targets Ther. 12, 6907–6915 (2019).
Guo, J. et al. A promising role of interferon regulatory factor 5 as an early warning biomarker for the development of human non-small cell lung cancer. Lung. Cancer 135, 47–55 (2019).
Yamashina, T. et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 74, 2698–2709 (2014).
Liu, Y. et al. Interferon regulatory factor 6 correlates with the progression of non-small cell lung cancer and can be regulated by miR-320. J. Pharm. Pharmacol. 73, 682–691 (2021).
Huang, L. et al. IRF7 and IFIT2 in mediating different hemorrhage outcomes for non-small cell lung cancer after bevacizumab treatment. J. Thorac. Dis. 15, 2022–2036 (2023).
Lai, Q. et al. Decitibine improve the efficiency of anti-PD-1 therapy via activating the response to IFN/PD-L1 signal of lung cancer cells. Oncogene 37, 2302–2312 (2018).
Suzuki, M. et al. Aberrant methylation and silencing of IRF8 expression in non-small cell lung cancer. Oncol. Lett. 8, 1025–1030 (2014).
Liang, J. et al. IRF8 induces senescence of lung cancer cells to exert its tumor suppressive function. Cell. Cycle 18, 3300–3312 (2019).
Li, L. et al. Integrated analysis of dysregulated long non-coding RNAs/microRNAs/mRNAs in metastasis of lung adenocarcinoma. J. Transl. Med. 16, 372 (2018).
Brunn, D. & Turkowski, K. Interferon Regulatory Factor 9 Promotes Lung Cancer Progression via Regulation of Versican. Cancers (Basel) 13, 208 (2021).
Cohen, S. et al. Interferon regulatory factor 1 is an independent predictor of platinum resistance and survival in high-grade serous ovarian carcinoma. Gynecol. Oncol. 134, 591–598 (2014).
Padmanabhan, S. et al. IFNγ-induced PD-L1 expression in ovarian cancer cells is regulated by JAK1, STAT1 and IRF1 signaling. Cell. Signal. 97, 110400 (2022).
Huang, S. L., Chang, T. C., Chao, C. C. K. & Sun, N. K. TLR4/IL-6/IRF1 signaling regulates androgen receptor expression: A potential therapeutic target to overcome taxol resistance in ovarian cancer. Biochem. Pharmacol. 186, 114456 (2021).
Pavan, S., Olivero, M., Corà, D. & Di Renzo, M. F. IRF-1 expression is induced by cisplatin in ovarian cancer cells and limits drug effectiveness. Eur. J. Cancer 49, 964–973 (2013).
Zhang, J. et al. Deubiquitinase USP35 restrains STING-mediated interferon signaling in ovarian cancer. Cell. Death. Differ. 28, 139–155 (2021).
Cornelison, R. et al. CX-5461 Treatment Leads to Cytosolic DNA-Mediated STING Activation in Ovarian Cancer. Cancers (Basel) 13, 5056 (2021).
Heimes, A. S. et al. A retrospective analysis of immunohistochemically determined IRF4 (interferon regulating factor 4) expression in a consecutive cohort of 114 ovarian cancer patients. Arch. Gynecol. Obstet. 299, 239–246 (2019).
Zhang, F. et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 10, 3974 (2019).
Hoffmann, M., Rak, A. & Ptak, A. Bisphenol A and its derivatives decrease expression of chemerin, which reverses its stimulatory action in ovarian cancer cells. Toxicol. Lett. 291, 61–69 (2018).
Schmitt, M. et al. Anti-Tumoral Effect of Chemerin on Ovarian Cancer Cell Lines Mediated by Activation of Interferon Alpha Response. Cancers (Basel) 14, 4108 (2022).
Tang, D. G. Understanding and targeting prostate cancer cell heterogeneity and plasticity. Semin. Cancer Biol. 82, 68–93 (2022).
Cheng, Y., Wang, D., Jiang, J. & Huang, W. Integrative analysis of AR-mediated transcriptional regulatory network reveals IRF1 as an inhibitor of prostate cancer progression. Prostate 80, 640–652 (2020).
Sonkusre, P. & Cameotra, S. S. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. J. Nanobiotechnology. 15, 43 (2017).
Bachmann, S. B. et al. DTX3L and ARTD9 inhibit IRF1 expression and mediate in cooperation with ARTD8 survival and proliferation of metastatic prostate cancer cells. Mol. Cancer 13, 125 (2014).
Wu, H. et al. Loss of a Negative Feedback Loop between IRF8 and AR Promotes Prostate Cancer Growth and Enzalutamide Resistance. Cancer Res. 80, 2927–2939 (2020).
Kneitz, B. et al. Survival in patients with high-risk prostate cancer is predicted by miR-221, which regulates proliferation, apoptosis, and invasion of prostate cancer cells by inhibiting IRF2 and SOCS3. Cancer Res. 74, 2591–2603 (2014).
Deveci Ozkan, A. & Kaleli, S. Anti-inflammatory effects of nobiletin on TLR4/TRIF/IRF3 and TLR9/IRF7 signaling pathways in prostate cancer cells. Immunopharmacol. Immunotoxicol. 42, 93–100 (2020).
Zhao, Y. et al. Overexpression of Interferon Regulatory Factor 7 (IRF7) Reduces Bone Metastasis of Prostate Cancer Cells in Mice. Oncol. Res. 25, 511–522 (2017).
Yu, S. J., Kim, H. S., Cho, S. W. & Sohn, J. IL-4 inhibits proliferation of renal carcinoma cells by increasing the expression of p21WAF1 and IRF-1. Exp. Mol. Med. 36, 372–379 (2004).
Tomita, Y. et al. Role of IRF-1 and caspase-7 in IFN-gamma enhancement of Fas-mediated apoptosis in ACHN renal cell carcinoma cells. Int. J. Cancer 104, 400–408 (2003).
Kong, S. K. et al. Dissection of PD-L1 promoter reveals differential transcriptional regulation of PD-L1 in VHL mutant clear cell renal cell carcinoma. Lab. Invest. 102, 352–362 (2022).
Wu, J. et al. Overexpression of IRF3 Predicts Poor Prognosis in Clear Cell Renal Cell Carcinoma. Int. J. Gen. Med. 14, 5675–5692 (2021).
Zeng, X., Li, L., Hu, Z. & Peng, D. Integrated Multi-Omics Analysis Identified PTPRG and CHL1 as Key Regulators of Immunophenotypes in Clear Cell Renal Cell Carcinoma(ccRCC). Front. Oncol. 12, 832027 (2022).
Wang, H. et al. ZNF692 promote proliferation through transcriptional repression of essential genes in clear cell renal carcinoma. Biochem. Biophys. Res. Commun. 671, 255–262 (2023).
Li, Z. et al. Decreased interferon regulatory factor 6 expression due to DNA hypermethylation predicts an unfavorable prognosis in clear cell renal cell carcinoma. J. Cancer 12, 6640–6655 (2021).
Wang, Y. et al. Gene Expression Microarray Data Meta-Analysis Identifies Candidate Genes and Molecular Mechanism Associated with Clear Cell Renal Cell Carcinoma. Cell. J. 22, 386–393 (2020).
Lin, L. & Cai, J. Circular RNA circ-EGLN3 promotes renal cell carcinoma proliferation and aggressiveness via miR-1299-mediated IRF7 activation. J. Cell. Biochem. 121, 4377–4385 (2020).
Muhitch, J. B. et al. Tumor-associated macrophage expression of interferon regulatory Factor-8 (IRF8) is a predictor of progression and patient survival in renal cell carcinoma. J. Immunother. Cancer 7, 155 (2019).
Zhang, Q. et al. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer Lett. 354, 227–234 (2014).
Nixon, B. G. et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity 55, 2044–2058.e2045 (2022).
Kamaraj, B., Al-Subaie, A. M., Ahmad, F. & Surapaneni, K. M. Effect of novel leukemia mutations (K75E & E222K) on interferon regulatory factor 1 and its interaction with DNA: insights from molecular dynamics simulations and docking studies. J. Biomol. Struct. Dyn. 39, 5235–5247 (2021).
Semmes, E. C. & Vijayakrishnan, J. Leveraging Genome and Phenome-Wide Association Studies to Investigate Genetic Risk of Acute Lymphoblastic Leukemia. Cancer Epidemiol. Biomark. Prev. 29, 1606–1614 (2020).
Manzella, L. et al. Roles of Interferon Regulatory Factors in Chronic Myeloid Leukemia. Curr. Cancer Drug. Targets 16, 594–605 (2016).
Mathew, N. R. et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 24, 282–291 (2018).
Wang, H. et al. Loss of IRF7 accelerates acute myeloid leukemia progression and induces VCAM1-VLA-4 mediated intracerebral invasion. Oncogene 41, 2303–2314 (2022).
Yang, X. et al. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology 7, e1412910 (2018).
Tian, W. L. et al. The IRF9-SIRT1-P53 axis is involved in the growth of human acute myeloid leukemia. Exp. Cell. Res. 365, 185–193 (2018).
Zhang, F. et al. IRF2-INPP4B axis participates in the development of acute myeloid leukemia by regulating cell growth and survival. Gene 627, 9–14 (2017).
Zhang, F. et al. IRF2-INPP4B-mediated autophagy suppresses apoptosis in acute myeloid leukemia cells. Biol. Res. 52, 11 (2019).
Zhang, F. et al. IRF2-INPP4B axis inhibits apoptosis of acute myeloid leukaemia cells via regulating T helper 1/2 cell differentiation. Cell. Biochem. Funct. 38, 582–590 (2020).
Zhang, F. et al. Exosomes derived from human bone marrow mesenchymal stem cells transfer miR-222-3p to suppress acute myeloid leukemia cell proliferation by targeting IRF2/INPP4B. Mol. Cell. Probes. 51, 101513 (2020).
Tian, W. L. et al. IRF3 is involved in human acute myeloid leukemia through regulating the expression of miR-155. Biochem. Biophys. Res. Commun. 478, 1130–1135 (2016).
Di Bernardo, M. C. et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat. Genet. 40, 1204–1210 (2008).
Wang, J. et al. Genome-Wide Association Analyses Identify Variants in IRF4 Associated With Acute Myeloid Leukemia and Myelodysplastic Syndrome Susceptibility. Front. Genet. 12, 554948 (2021).
Maffei, R., Fiorcari, S., Benatti, S. & Atene, C. G. IRF4 modulates the response to BCR activation in chronic lymphocytic leukemia regulating IKAROS and SYK. 35, 1330-1343, (2021).
Asslaber, D. et al. B-cell-specific IRF4 deletion accelerates chronic lymphocytic leukemia development by enhanced tumor immune evasion. Blood 134, 1717–1729 (2019).
Zhang, Y. & Zeng, X. Correlation of the transcription factors IRF4 and BACH2 with the abnormal NFATC1 expression in T cells from chronic myeloid leukemia patients. Hematology 27, 523–529 (2022).
Tarantini, F. et al. IRF4 Gene Expression on the Trail of Molecular Response: Looking at Chronic Myeloid Leukemia from Another Perspective. Acta Haematol. 146, 37–43 (2023).
Pang, S. H. et al. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia 30, 1375–1387 (2016).
So, A. Y. et al. Dual mechanisms by which miR-125b represses IRF4 to induce myeloid and B-cell leukemias. Blood 124, 1502–1512 (2014).
Wong, R. W. J. et al. Feed-forward regulatory loop driven by IRF4 and NF-κB in adult T-cell leukemia/lymphoma. Blood 135, 934–947 (2020).
Sakamoto, H. & Ando, K. Alvocidib inhibits IRF4 expression via super-enhancer suppression and adult T-cell leukemia/lymphoma cell growth. Cancer Sci. 113, 4092–4103 (2022).
Watanabe, T. et al. The transcription factor IRF8 counteracts BCR-ABL to rescue dendritic cell development in chronic myelogenous leukemia. Cancer Res. 73, 6642–6653 (2013).
Hu, X. et al. IRF8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer Res. 71, 2882–2891 (2011).
Scheller, M. et al. Cross talk between Wnt/β-catenin and Irf8 in leukemia progression and drug resistance. J. Exp. Med. 210, 2239–2256 (2013).
Gaillard, C. et al. Identification of IRF8 as a potent tumor suppressor in murine acute promyelocytic leukemia. Blood. Adv. 2, 2462–2466 (2018).
Zhou, Y. et al. Silencing of IRF8 Mediated by m6A Modification Promotes the Progression of T-Cell Acute Lymphoblastic Leukemia. Adv. Sci. (Weinh.). 10, e2201724 (2023).
Liss, F. et al. IRF8 Is an AML-Specific Susceptibility Factor That Regulates Signaling Pathways and Proliferation of AML Cells. Cancers (Basel) 13, 764 (2021).
Zhuang, H. et al. Loss of IRF8 inhibits the growth of acute myeloid leukemia cells. Ann. Hematol. 102, 1063–1072 (2023).
Cao, Z. et al. ZMYND8-regulated IRF8 transcription axis is an acute myeloid leukemia dependency. Mol. Cell. 81, 3604–3622.e3610 (2021).
Pingul, B. Y. et al. Dissection of the MEF2D-IRF8 transcriptional circuit dependency in acute myeloid leukemia. iScience 25, 105139 (2022).
Wiseman, D. H. et al. Chronic myelomonocytic leukaemia stem cell transcriptomes anticipate disease morphology and outcome. EBioMedicine 58, 102904 (2020).
Wang, Q. & Lin, Z. RARγ activation sensitizes human myeloma cells to carfilzomib treatment through the OAS-RNase L innate immune pathway. Blood 139, 59–72 (2022).
Xing, L., Wang, S. & Liu, J. BCMA-Specific ADC MEDI2228 and Daratumumab Induce Synergistic Myeloma Cytotoxicity via IFN-Driven Immune Responses and Enhanced CD38 Expression. Clin. Cancer Res. 27, 5376–5388 (2021).
Tsubaki, M. et al. Macrophage inflammatory protein-1α induces osteoclast formation by activation of the MEK/ERK/c-Fos pathway and inhibition of the p38MAPK/IRF-3/IFN-β pathway. J. Cell. Biochem. 111, 1661–1672 (2010).
Liu, H., He, J. & Bagheri-Yarmand, R. Osteocyte CIITA aggravates osteolytic bone lesions in myeloma. Nat. Commun. 13, 3684 (2022).
Fedele, P. L., Liao, Y., Gong, J. N. & Yao, Y. The transcription factor IRF4 represses proapoptotic BMF and BIM to licence multiple myeloma survival. Leukemia 35, 2114–2118 (2021).
Katsarou, A., Trasanidis, N., Ponnusamy, K. & Kostopoulos, I. V. MAF functions as a pioneer transcription factor that initiates and sustains myelomagenesis. Blood. Adv. 7, 6395–6410 (2023).
Du, L., Liu, W., Pichiorri, F. & Rosen, S. T. SUMOylation inhibition enhances multiple myeloma sensitivity to lenalidomide. Cancer Gene. Ther. 30, 567–574 (2023).
Mougiakakos, D., Bach, C. & Böttcher, M. The IKZF1-IRF4/IRF5 Axis Controls Polarization of Myeloma-Associated Macrophages. Cancer Immunol. Res. 9, 265–278 (2021).
Mondala, P. K. et al. Selective antisense oligonucleotide inhibition of human IRF4 prevents malignant myeloma regeneration via cell cycle disruption. Cell. Stem. Cell. 28, 623–636.e629 (2021).
Morelli, E. et al. Selective targeting of IRF4 by synthetic microRNA-125b-5p mimics induces anti-multiple myeloma activity in vitro and in vivo. Leukemia 29, 2173–2183 (2015).
Wang, Q. S. et al. Identification of Immune-Related Genes for Risk Stratification in Multiple Myeloma Based on Whole Bone Marrow Gene Expression Profiling. Front. Genet. 13, 897886 (2022).
Waller, R. G. et al. Sequencing at lymphoid neoplasm susceptibility loci maps six myeloma risk genes. Hum. Mol. Genet. 30, 1142–1153 (2021).
Liu, R. et al. m(6)A reader hnRNPA2B1 drives multiple myeloma osteolytic bone disease. Theranostics 12, 7760–7774 (2022).
Giannouli, S. et al. Autoimmune manifestations in human myelodysplasia: a positive correlation with interferon regulatory factor-1 (IRF-1) expression. Ann. Rheum. Dis. 63, 578–582 (2004).
Pinheiro, R. F., Metze, K., Silva, M. R. & Chauffaille Mde, L. The ambiguous role of interferon regulatory factor-1 (IRF-1) immunoexpression in myelodysplastic syndrome. Leuk. Res. 33, 1308–1312 (2009).
de Sousa, J. C. et al. Dysregulation of interferon regulatory genes reinforces the concept of chronic immune response in myelodysplastic syndrome pathogenesis. Hematol. Oncol. 37, 523–526 (2019).
de Oliveira, R. T. G. et al. ERVs-TLR3-IRF axis is linked to myelodysplastic syndrome pathogenesis. Med. Oncol. 38, 27 (2021).
Le, Y. Screening and identification of key candidate genes and pathways in myelodysplastic syndrome by bioinformatic analysis. PeerJ 7, e8162 (2019).
Wang, S. et al. CTLA-4 blockade induces tumor pyroptosis via CD8(+) T cells in head and neck squamous cell carcinoma. Mol. Ther. 31, 2154–2168 (2023).
Li, X. & Yang, W. IRF2-induced Claudin-7 suppresses cell proliferation, invasion and migration of oral squamous cell carcinoma. Exp. Ther. Med. 23, 7 (2022).
Jin, S. et al. The m6A demethylase ALKBH5 promotes tumor progression by inhibiting RIG-I expression and interferon alpha production through the IKKε/TBK1/IRF3 pathway in head and neck squamous cell carcinoma. Mol. Cancer 21, 97 (2022).
Yan, Y., Gauthier, M. A. & Malik, A. The NOTCH-RIPK4-IRF6-ELOVL4 Axis Suppresses Squamous Cell Carcinoma. Cancers (Basel) 15, 737 (2023).
Ke, L. et al. Nasopharyngeal carcinoma super-enhancer-driven ETV6 correlates with prognosis. Proc. Natl Acad. Sci. Usa. 114, 9683–9688 (2017).
Qi, C. L. et al. The IRF2/CENP-N/AKT signaling axis promotes proliferation, cell cycling and apoptosis resistance in nasopharyngeal carcinoma cells by increasing aerobic glycolysis. J. Exp. Clin. Cancer Res. 40, 390 (2021).
Ge, J. et al. Epstein-Barr Virus-Encoded Circular RNA CircBART2.2 Promotes Immune Escape of Nasopharyngeal Carcinoma by Regulating PD-L1. Cancer Res. 81, 5074–5088 (2021).
Wan, P., Zhang, J., Du, Q. & Geller, D. A. The clinical significance and biological function of interferon regulatory factor 1 in cholangiocarcinoma. Biomed. Pharmacother. 97, 771–777 (2018).
Wan, P. et al. miR-383 promotes cholangiocarcinoma cell proliferation, migration, and invasion through targeting IRF1. J. Cell. Biochem. 119, 9720–9729 (2018).
Wei, C. X. et al. IRF4-induced upregulation of lncRNA SOX2-OT promotes cell proliferation and metastasis in cholangiocarcinoma by regulating SOX2 and PI3K/AKT signaling. Eur. Rev. Med. Pharmacol. Sci. 22, 8169–8178 (2018).
Meyer-Schaller, N. et al. A dual role of Irf1 in maintaining epithelial identity but also enabling EMT and metastasis formation of breast cancer cells. Oncogene 39, 4728–4740 (2020).
Wu, B. et al. UBR5 promotes tumor immune evasion through enhancing IFN-γ-induced PDL1 transcription in triple negative breast cancer. Theranostics 12, 5086–5102 (2022).
Schwartz-Roberts, J. L. et al. Interferon regulatory factor-1 signaling regulates the switch between autophagy and apoptosis to determine breast cancer cell fate. Cancer Res. 75, 1046–1055 (2015).
Armstrong, M. J. et al. IRF-1 inhibits NF-κB activity, suppresses TRAF2 and cIAP1 and induces breast cancer cell specific growth inhibition. Cancer Biol. Ther. 16, 1029–1041 (2015).
Luker, K. E., Pica, C. M., Schreiber, R. D. & Piwnica-Worms, D. Overexpression of IRF9 confers resistance to antimicrotubule agents in breast cancer cells. Cancer Res. 61, 6540–6547 (2001).
Brockwell, N. K. et al. Tumor inherent interferon regulators as biomarkers of long-term chemotherapeutic response in TNBC. Npj. Precis. Oncol. 3, 21 (2019).
Bernardo, A. R., Cosgaya, J. M., Aranda, A. & Jiménez-Lara, A. M. Pro-apoptotic signaling induced by Retinoic acid and dsRNA is under the control of Interferon Regulatory Factor-3 in breast cancer cells. Apoptosis 22, 920–932 (2017).
Lu, X. et al. Anti-triple-negative breast cancer metastasis efficacy and molecular mechanism of the STING agonist for innate immune pathway. Ann. Med. 55, 2210845 (2023).
Pantelidou, C. & Sonzogni, O. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 9, 722–737 (2019).
Vernier, M. et al. Inhibition of DNMT1 and ERRα crosstalk suppresses breast cancer via derepression of IRF4. Oncogene 39, 6406–6420 (2020).
Heimes, A. S. et al. Prognostic significance of interferon regulating factor 4 (IRF4) in node-negative breast cancer. J. Cancer Res. Clin. Oncol. 143, 1123–1131 (2017).
Pimenta, E. M. & Barnes, B. J. A conserved region within interferon regulatory factor 5 controls breast cancer cell migration through a cytoplasmic and transcription-independent mechanism. Mol. Cancer 14, 32 (2015).
Bi, X. et al. Loss of interferon regulatory factor 5 (IRF5) expression in human ductal carcinoma correlates with disease stage and contributes to metastasis. Breast Cancer Res. 13, R111 (2011).
Xu, H. F. et al. Candidate tumor suppressor gene IRF6 is involved in human breast cancer pathogenesis via modulating PI3K-regulatory subunit PIK3R2 expression. Cancer Manag. Res. 11, 5557–5572 (2019).
Liu, X. & Chipurupalli, S. ErbB2/Her2-dependent downregulation of a cell death-promoting protein BLNK in breast cancer cells is required for 3D breast tumor growth. Cell. Death. Dis. 13, 687 (2022).
Khan, I. A. et al. ErbB2-driven downregulation of the transcription factor Irf6 in breast epithelial cells is required for their 3D growth. Breast Cancer Res. 20, 151 (2018).
de Mingo Pulido, Á. et al. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell. 33, 60–74.e66 (2018).
Cohen, P. A., Jhingran, A., Oaknin, A. & Denny, L. Cervical cancer. Lancet 393, 169–182 (2019).
Rho, S. B., Lee, S. H., Byun, H. J. & Kim, B. R. IRF-1 Inhibits Angiogenic Activity of HPV16 E6 Oncoprotein in Cervical Cancer. Int. J. Mol. Sci. 21, 7622 (2020).
Walch-Rückheim, B. et al. STAT3/IRF1 Pathway Activation Sensitizes Cervical Cancer Cells to Chemotherapeutic Drugs. Cancer Res. 76, 3872–3883 (2016).
Lee, S. H. et al. IFN-gamma/IRF-1-induced p27kip1 down-regulates telomerase activity and human telomerase reverse transcriptase expression in human cervical cancer. Febs. Lett. 579, 1027–1033 (2005).
Deng, Z. M., Dai, F. F., Zhou, Q. & Cheng, Y. X. Hsa_circ_0000301 facilitates the progression of cervical cancer by targeting miR-1228-3p/IRF4 Axis. Bmc. Cancer 21, 583 (2021).
Ren, Y., Dong, J. & He, P. miR-587 promotes cervical cancer by repressing interferon regulatory factor 6. J. Gene. Med. 22, e3257 (2020).
Kuroboshi, H. et al. Interferon regulatory factor-1 expression in human uterine endometrial carcinoma. Gynecol. Oncol. 91, 354–358 (2003).
Gao, K. et al. SPOP mutations promote tumor immune escape in endometrial cancer via the IRF1-PD-L1 axis. Cell. Death. Differ. 30, 475–487 (2023).
Zeng, X. et al. IL6 Induces mtDNA Leakage to Affect the Immune Escape of Endometrial Carcinoma via cGAS-STING. J. Immunol. Res. 2022, 3815853 (2022).
Nakamura, K. et al. ADAR1 and AZIN1 RNA editing function as an oncogene and contributes to immortalization in endometrial cancer. Gynecol. Oncol. 166, 326–333 (2022).
Ghislat, G. et al. NF-κB-dependent IRF1 activation programs cDC1 dendritic cells to drive antitumor immunity. Sci. Immunol. 6, eabg3570 (2021).
Zhou, S. et al. IRF1 expression might be a biomarker of CD8+ T cell infiltration in cutaneous melanoma. Expert. Rev. Clin. Immunol. 18, 1319–1327 (2022).
Mirjačić Martinović, K. et al. Decreased expression of pSTAT, IRF-1 and DAP10 signalling molecules in peripheral blood lymphocytes of patients with metastatic melanoma. J. Clin. Pathol. 69, 300–306 (2016).
Yokoyama, S. et al. SOX10 Regulates Melanoma Immunogenicity through an IRF4-IRF1 Axis. Cancer Res. 81, 6131–6141 (2021).
Wang, W. et al. Upregulation of PD-L1 via HMGB1-Activated IRF3 and NF-κB Contributes to UV Radiation-Induced Immune Suppression. Cancer Res. 79, 2909–2922 (2019).
Occhigrossi, L. & D’Eletto, M. Transglutaminase type 2-dependent crosslinking of IRF3 in dying melanoma cells. Cell. Death. Discov. 8, 498 (2022).
Musick, M. & Yu, X. Immunotherapeutic effects of intratumorally injected Zymosan-Adenovirus conjugates encoding constant active IRF3 in a melanoma mouse model. Immunol. Res. 71, 197–212 (2023).
Humblin, E. et al. IRF8-dependent molecular complexes control the Th9 transcriptional program. Nat. Commun. 8, 2085 (2017).
Wang, D. et al. Increased IRF9-STAT2 Signaling Leads to Adaptive Resistance toward Targeted Therapy in Melanoma by Restraining GSDME-Dependent Pyroptosis. J. Invest. Dermatol. 142, 2476–2487.e2479 (2022).
Chu, Z., Gu, L., Hu, Y. & Zhang, X. STAG2 regulates interferon signaling in melanoma via enhancer loop reprogramming. Nat. Commun. 13, 1859 (2022).
Sancéau, J., Hiscott, J., Delattre, O. & Wietzerbin, J. IFN-beta induces serine phosphorylation of Stat-1 in Ewing’s sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene 19, 3372–3383 (2000).
Ma, Z. et al. MiR-134, Mediated by IRF1, Suppresses Tumorigenesis and Progression by Targeting VEGFA and MYCN in Osteosarcoma. Anticancer. Agents Med. Chem. 20, 1197–1208 (2020).
Zhang, X. et al. The CtBP1-HDAC1/2-IRF1 transcriptional complex represses the expression of the long noncoding RNA GAS5 in human osteosarcoma cells. Int. J. Biol. Sci. 15, 1460–1471 (2019).
Cheng, J. P. et al. miR-4295 promotes cell proliferation, migration and invasion of osteosarcoma through targeting interferon regulatory factor 1. Oncol. Lett. 20, 260 (2020).
Lu, C. et al. miR-18a-5p promotes cell invasion and migration of osteosarcoma by directly targeting IRF2. Oncol. Lett. 16, 3150–3156 (2018).
Wu, W. et al. SGLT2 inhibitor activates the STING/IRF3/IFN-β pathway and induces immune infiltration in osteosarcoma. Cell. Death. Dis. 13, 523 (2022).
Li, Z. et al. IRF7 inhibits the Warburg effect via transcriptional suppression of PKM2 in osteosarcoma. Int. J. Biol. Sci. 18, 30–42 (2022).
Li, Z. et al. Erratum: IRF7 inhibits the Warburg effect via transcriptional suppression of PKM2 in osteosarcoma: Erratum. Int. J. Biol. Sci. 18, 6229–6230 (2022).
Ni, X. & Wu, W. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci. Adv. 8, eabl5165 (2022).
Tong, S. et al. IRF2-ferroptosis related gene is associated with prognosis and EMT in gliomas. Transl. Oncol. 26, 101544 (2022).
Tarassishin, L. & Lee, S. C. Interferon regulatory factor 3 alters glioma inflammatory and invasive properties. J. Neurooncol. 113, 185–194 (2013).
Pattwell, S. S. & Holland, E. C. Putting Glioblastoma in Its Place: IRF3 Inhibits Invasion. Trends Mol. Med. 23, 773–776 (2017).
Pencheva, N. et al. Identification of a Druggable Pathway Controlling Glioblastoma Invasiveness. Cell. Rep. 20, 48–60 (2017).
Yang, T. et al. A BRD4 PROTAC nanodrug for glioma therapy via the intervention of tumor cells proliferation, apoptosis and M2 macrophages polarization. Acta Pharm. Sin. B. 12, 2658–2671 (2022).
Li, C., Guan, N. & Liu, F. T7 peptide-decorated exosome-based nanocarrier system for delivery of Galectin-9 siRNA to stimulate macrophage repolarization in glioblastoma. J. Neurooncol. 162, 93–108 (2023).
Shah, D. & Comba, A. A novel miR1983-TLR7-IFNβ circuit licenses NK cells to kill glioma cells, and is under the control of galectin-1. Oncoimmunology 10, 1939601 (2021).
Tang, R. et al. Opposite effects of interferon regulatory factor 1 and osteopontin on the apoptosis of epithelial cells induced by TNF-α in inflammatory bowel disease. Inflamm. Bowel. Dis. 20, 1950–1961 (2014).
Tan, G. et al. An IRF1-dependent Pathway of TNFα-induced Shedding in Intestinal Epithelial Cells. J. Crohns. Colitis 16, 133–142 (2022).
Xu, H. & Fu, J. TNF-α Enhances the Therapeutic Effects of MenSC-Derived Small Extracellular Vesicles on Inflammatory Bowel Disease through Macrophage Polarization by miR-24-3p. Stem. Cells Int. 2023, 2988907 (2023).
Wang, K. W. et al. Enhanced susceptibility to chemically induced colitis caused by excessive endosomal TLR signaling in LRBA-deficient mice. Proc. Natl Acad. Sci. USA 116, 11380–11389 (2019).
Chiriac, M. T. et al. Activation of Epithelial Signal Transducer and Activator of Transcription 1 by Interleukin 28 Controls Mucosal Healing in Mice With Colitis and Is Increased in Mucosa of Patients With Inflammatory Bowel Disease. Gastroenterology 153, 123–138.e128 (2017).
Buchele, V. et al. Th17 Cell-Mediated Colitis Is Positively Regulated by Interferon Regulatory Factor 4 in a T Cell-Extrinsic Manner. Front. Immunol. 11, 590893 (2020).
Zhu, X. et al. 1,25‑Dihydroxyvitamin D regulates macrophage polarization and ameliorates experimental inflammatory bowel disease by suppressing miR-125b. Int. Immunopharmacol. 67, 106–118 (2019).
Zhu, W. et al. Baicalin ameliorates experimental inflammatory bowel disease through polarization of macrophages to an M2 phenotype. Int. Immunopharmacol. 35, 119–126 (2016).
Yan, J. et al. T Cell-Intrinsic IRF5 Regulates T Cell Signaling, Migration, and Differentiation and Promotes Intestinal Inflammation. Cell. Rep. 31, 107820 (2020).
Kayama, H. et al. Heme ameliorates dextran sodium sulfate-induced colitis through providing intestinal macrophages with noninflammatory profiles. Proc. Natl Acad. Sci. USA 115, 8418–8423 (2018).
Ryzhakov, G., Almuttaqi, H. & Corbin, A. L. Defactinib inhibits PYK2 phosphorylation of IRF5 and reduces intestinal inflammation. Nat. Commun. 12, 6702 (2021).
Lu, J. et al. Thalidomide Attenuates Colitis and Is Associated with the Suppression of M1 Macrophage Polarization by Targeting the Transcription Factor IRF5. Dig. Dis. Sci. 66, 3803–3812 (2021).
Lin, Z. et al. microRNA-144/451 decreases dendritic cell bioactivity via targeting interferon-regulatory factor 5 to limit DSS-induced colitis. Front. Immunol. 13, 928593 (2022).
Zhang, R. et al. T follicular helper cells restricted by IRF8 contribute to T cell-mediated inflammation. J. Autoimmun. 96, 113–122 (2019).
Veiga, N. et al. Leukocyte-specific siRNA delivery revealing IRF8 as a potential anti-inflammatory target. J. Control. Release 313, 33–41 (2019).
Landgraf-Rauf, K. et al. IRF-1 SNPs influence the risk for childhood allergic asthma: A critical role for pro-inflammatory immune regulation. Pediatr. Allergy Immunol. 29, 34–41 (2018).
Kumar, A. et al. Genetic association of key Th1/Th2 pathway candidate genes, IRF2, IL6, IFNGR2, STAT4 and IL4RA, with atopic asthma in the Indian population. J. Hum. Genet. 60, 443–448 (2015).
Ozasa, K. et al. Cyclic GMP-AMP Triggers Asthma in an IL-33-Dependent Manner That Is Blocked by Amlexanox, a TBK1 Inhibitor. Front. Immunol. 10, 2212 (2019).
Marichal, T. et al. Interferon response factor 3 is essential for house dust mite-induced airway allergy. J. Allergy Clin. Immunol. 126, 836–844.e813 (2010).
He, J. et al. IRF-7 Is a Critical Regulator of Type 2 Innate Lymphoid Cells in Allergic Airway Inflammation. Cell. Rep. 29, 2718–2730.e2716 (2019).
Park, S. J. et al. Tilianin attenuates HDM-induced allergic asthma by suppressing Th2-immune responses via downregulation of IRF4 in dendritic cells. Phytomedicine 80, 153392 (2021).
Xia, L. et al. lnc-BAZ2B promotes M2 macrophage activation and inflammation in children with asthma through stabilizing BAZ2B pre-mRNA. J. Allergy Clin. Immunol. 147, 921–932.e929 (2021).
Corrigendum. J. Allergy. Clin. Immunol. 149, 1134-1135, (2022).
Hwang, S. S. et al. RHS6 coordinately regulates the Th2 cytokine genes by recruiting GATA3, SATB1, and IRF4. Allergy 72, 772–782 (2017).
Kim, S. B. et al. Anthriscus sylvestris root extract reduces allergic lung inflammation by regulating interferon regulatory factor 4-mediated Th2 cell activation. J. Ethnopharmacol. 232, 165–175 (2019).
Staudt, V. et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 33, 192–202 (2010).
Oriss, T. B. et al. IRF5 distinguishes severe asthma in humans and drives Th1 phenotype and airway hyperreactivity in mice. JCI. Insight. 2, e91019 (2017).
Swindell, W. R. et al. Psoriasis drug development and GWAS interpretation through in silico analysis of transcription factor binding sites. Clin. Transl. Med. 4, 13 (2015).
Kuai, L. et al. Celastrol Attenuates Psoriasiform Inflammation by Targeting the IRF1/GSTM3 Axis. J. Invest. Dermatol. 142, 2281–2285.e2211 (2022).
Lin, S. H. et al. Treatment with TNF-α inhibitor rectifies M1 macrophage polarization from blood CD14+ monocytes in patients with psoriasis independent of STAT1 and IRF-1 activation. J. Dermatol. Sci. 91, 276–284 (2018).
Parkinson, J. et al. Variation at the IRF2 gene and susceptibility to psoriasis in chromosome 4q-linked families. J. Invest. Dermatol. 122, 640–643 (2004).
Foerster, J. et al. Evaluation of the IRF-2 gene as a candidate for PSORS3. J. Invest. Dermatol. 122, 61–64 (2004).
van der Fits, L. et al. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered. J. Invest. Dermatol. 122, 51–60 (2004).
Kawaguchi, M. et al. IRF-2 haploinsufficiency causes enhanced imiquimod-induced psoriasis-like skin inflammation. J. Dermatol. Sci. 90, 35–45 (2018).
Xiaohong, L. et al. Activation of the STING-IRF3 pathway involved in psoriasis with diabetes mellitus. J. Cell. Mol. Med. 26, 2139–2151 (2022).
Ni, A. et al. Expression of IRF-4 and IBP in skin lesions of patients with psoriasis vulgaris. J. Huazhong. Univ. Sci. Technol. Med. Sci. 32, 287–290 (2012).
Cai, Y. et al. Differential Roles of the mTOR-STAT3 Signaling in Dermal γδ T Cell Effector Function in Skin Inflammation. Cell. Rep. 27, 3034–3048.e3035 (2019).
Nakao, M. & Miyagaki, T. Exacerbated Imiquimod-Induced Psoriasis-Like Skin Inflammation in IRF5-Deficient Mice. Int. J. Mol. Sci. 21, 3681 (2020).
Raposo, R. A. et al. Antiviral gene expression in psoriasis. J. Eur. Acad. Dermatol. Venereol. 29, 1951–1957 (2015).
Jin, L. et al. A BET inhibitor, NHWD-870, can downregulate dendritic cells maturation via the IRF7-mediated signaling pathway to ameliorate imiquimod-induced psoriasis-like murine skin inflammation. Eur. J. Pharmacol. 968, 176382 (2024).
Zhou, B. et al. Zdhhc2 Is Essential for Plasmacytoid Dendritic Cells Mediated Inflammatory Response in Psoriasis. Front. Immunol. 11, 607442 (2020).
Morell, M. et al. SIDT1 plays a key role in type I IFN responses to nucleic acids in plasmacytoid dendritic cells and mediates the pathogenesis of an imiquimod-induced psoriasis model. EBioMedicine 76, 103808 (2022).
Su, W. et al. Exploring the Pathogenesis of Psoriasis Complicated With Atherosclerosis via Microarray Data Analysis. Front. Immunol. 12, 667690 (2021).
Zolotarenko, A. et al. Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis. Exp. Mol. Med. 48, e268 (2016).
Gao, P. S. et al. Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J. Invest. Dermatol. 132, 650–657 (2012).
Li, C., Lu, Y. & Han, X. Identification of Effective Diagnostic Biomarkers and Immune Cell Infiltration in Atopic Dermatitis by Comprehensive Bioinformatics Analysis. Front. Mol. Biosci. 9, 917077 (2022).
Leung, Y. T. et al. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics 10, 191–199 (2015).
Corrigendum. Epigenetics 10, 891, (2015).
Zhang, Z. et al. Interferon regulatory factor 1 marks activated genes and can induce target gene expression in systemic lupus erythematosus. Arthritis Rheumatol. 67, 785–796 (2015).
Misidentified Band in Figure in the Article by Zhang et al (Arthritis Rheumatol, March 2015). Arthritis. Rheumatol. 67, 2481, (2015).
Liu, J., Berthier, C. C. & Kahlenberg, J. M. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol. 69, 1840–1849 (2017).
Chen, J. et al. HDAC1 potentiates CD4 + T cell activation by inhibiting miR-124 and promoting IRF1 in systemic lupus erythematosus. Cell. Immunol. 362, 104284 (2021).
Han, X. et al. MicroRNA-130b Ameliorates Murine Lupus Nephritis Through Targeting the Type I Interferon Pathway on Renal Mesangial Cells. Arthritis Rheumatol. 68, 2232–2243 (2016).
Zhang, F. et al. Independent Replication on Genome-Wide Association Study Signals Identifies IRF3 as a Novel Locus for Systemic Lupus Erythematosus. Front. Genet. 11, 600 (2020).
Xu, Z. Q. et al. CircELK4 Contributes to Lupus Nephritis by Acting as a miR-27b-3p Sponge to Regulate STING/IRF3/IFN-I Signaling. 44, 2106-2119, (2021).
Yoo, E. J. et al. Macrophage transcription factor TonEBP promotes systemic lupus erythematosus and kidney injury via damage-induced signaling pathways. Kidney Int. 104, 163–180 (2023).
Zheng, X. et al. AKT2 reduces IFNβ1 production to modulate antiviral responses and systemic lupus erythematosus. Embo. J. 41, e108016 (2022).
Faridi, M. H. et al. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J. Clin. Invest. 127, 1271–1283 (2017).
Yao, M. et al. Identification of Molecular Markers Associated With the Pathophysiology and Treatment of Lupus Nephritis Based on Integrated Transcriptome Analysis. Front. Genet. 11, 583629 (2020).
Luque, A. et al. Noncanonical immunomodulatory activity of complement regulator C4BP(β-) limits the development of lupus nephritis. Kidney Int. 97, 551–566 (2020).
Coit, P. et al. Hypomethylation of miR-17-92 cluster in lupus T cells and no significant role for genetic factors in the lupus-associated DNA methylation signature. Ann. Rheum. Dis. 81, 1428–1437 (2022).
Miyagawa, F. & Tagaya, Y. Essential Requirement for IFN Regulatory Factor 7 in Autoantibody Production but Not Development of Nephritis in Murine Lupus. J. Immunol. 197, 2167–2176 (2016).
Chandrasekaran, U. et al. Regulation of Effector Treg Cells in Murine Lupus. Arthritis Rheumatol. 68, 1454–1466 (2016).
Lech, M. et al. IRF4 deficiency abrogates lupus nephritis despite enhancing systemic cytokine production. J. Am. Soc. Nephrol. 22, 1443–1452 (2011).
Graham, R. R. et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat. Genet. 38, 550–555 (2006).
Graham, R. R. et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc. Natl Acad. Sci. Usa. 104, 6758–6763 (2007).
Barnes, B. J. Genetic Versus Non-genetic Drivers of SLE: Implications of IRF5 Dysregulation in Both Roads Leading to SLE. Curr. Rheumatol. Rep. 21, 2 (2019).
Hou, G. et al. Integrative Functional Genomics Identifies Systemic Lupus Erythematosus Causal Genetic Variant in the IRF5 Risk Locus. Arthritis Rheumatol. 75, 574–585 (2023).
Li, D. et al. IRF5 genetic risk variants drive myeloid-specific IRF5 hyperactivation and presymptomatic SLE. JCI. Insight. 5, e124020 (2020).
De, S. et al. B Cell-Intrinsic Role for IRF5 in TLR9/BCR-Induced Human B Cell Activation, Proliferation, and Plasmablast Differentiation. Front. Immunol. 8, 1938 (2017).
Pellerin, A. et al. Monoallelic IRF5 deficiency in B cells prevents murine lupus. JCI. Insight. 6, (2021).
Ban, T. et al. Genetic and chemical inhibition of IRF5 suppresses pre-existing mouse lupus-like disease. Nat. Commun. 12, 4379 (2021).
Song, S. et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J. Clin. Invest. 130, 6700–6717 (2020).
Zhou, T., Zhu, X., Ye, Z. & Wang, Y. F. Lupus enhancer risk variant causes dysregulation of IRF8 through cooperative lncRNA and DNA methylation machinery. Nat. Commun. 13, 1855 (2022).
Ji, J. et al. Myeloid-derived suppressor cells contribute to systemic lupus erythaematosus by regulating differentiation of Th17 cells and Tregs. Clin. Sci. (Lond.). 130, 1453–1467 (2016).
Smith, S. et al. MicroRNA-302d targets IRF9 to regulate the IFN-induced gene expression in SLE. J. Autoimmun. 79, 105–111 (2017).
Deng, Y. et al. Expression characteristics of interferon-stimulated genes and possible regulatory mechanisms in lupus patients using transcriptomics analyses. EBioMedicine 70, 103477 (2021).
Annibali, V. et al. Analysis of coding and non-coding transcriptome of peripheral B cells reveals an altered interferon response factor (IRF)-1 pathway in multiple sclerosis patients. J. Neuroimmunol. 324, 165–171 (2018).
Kortam, M. A. et al. MAGI2-AS3 and miR-374b-5p as Putative Regulators of Multiple Sclerosis via Modulating the PTEN/AKT/IRF-3/IFN-β Axis: New Clinical Insights. Acs. Chem. Neurosci. 14, 1107–1118 (2023).
Khaw, Y. M. & Anwar, S. Estrogen receptor alpha signaling in dendritic cells modulates autoimmune disease phenotype in mice. Embo. Rep. 24, e54228 (2023).
Sha, Y. & Markovic-Plese, S. Activated IL-1RI Signaling Pathway Induces Th17 Cell Differentiation via Interferon Regulatory Factor 4 Signaling in Patients with Relapsing-Remitting Multiple Sclerosis. Front. Immunol. 7, 543 (2016).
Zhao, M., Sun, D., Guan, Y. & Wang, Z. Disulfiram and Diphenhydramine Hydrochloride Upregulate miR-30a to Suppress IL-17-Associated Autoimmune Inflammation. J. Neurosci. 36, 9253–9266 (2016).
Kiselev, I. et al. Genetic differences between primary progressive and relapsing-remitting multiple sclerosis: The impact of immune-related genes variability. Mult. Scler. Relat. Disord. 29, 130–136 (2019).
Kowalec, K. & Wright, G. E. B. Common variation near IRF6 is associated with IFN-β-induced liver injury in multiple sclerosis. Nat. Genet. 50, 1081–1085 (2018).
Hoppmann, N. et al. New candidates for CD4 T cell pathogenicity in experimental neuroinflammation and multiple sclerosis. Brain 138, 902–917 (2015).
Kotelnikova, E. et al. MAPK pathway and B cells overactivation in multiple sclerosis revealed by phosphoproteomics and genomic analysis. Proc. Natl Acad. Sci. USA 116, 9671–9676 (2019).
Sellebjerg, F. et al. Prediction of response to interferon therapy in multiple sclerosis. Acta Neurol. Scand. 130, 268–275 (2014).
Yoshida, Y. et al. The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation. Immunity 40, 187–198 (2014).
Wei, Y. et al. Aryl hydrocarbon receptor activation drives polymorphonuclear myeloid-derived suppressor cell response and efficiently attenuates experimental Sjögren’s syndrome. Cell. Mol. Immunol. 19, 1361–1372 (2022).
Xiao, F. et al. Artesunate suppresses Th17 response via inhibiting IRF4-mediated glycolysis and ameliorates Sjögren’s syndrome. Signal. Transduct. Target Ther. 7, 274 (2022).
Nordmark, G. et al. Additive effects of the major risk alleles of IRF5 and STAT4 in primary Sjögren’s syndrome. Genes. Immun. 10, 68–76 (2009).
Ortíz-Fernández, L., Martín, J. & Alarcón-Riquelme, M. E. A Summary on the Genetics of Systemic Lupus Erythematosus, Rheumatoid Arthritis, Systemic Sclerosis, and Sjögren’s Syndrome. Clin. Rev. Allergy Immunol. 64, 392–411 (2023).
Bonelli, M. et al. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes: JAKinibs suppress the interferon response in RA-FLSs. Exp. Mol. Med. 51, 1–11 (2019).
Du, Y. et al. Regulation of type I interferon signature by VGLL3 in the fibroblast-like synoviocytes of rheumatoid arthritis patients via targeting the Hippo pathway. Arthritis Res. Ther. 24, 188 (2022).
van Hamburg, J. P. & Tas, S. W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 87, 69–81 (2018).
López-Isac, E. et al. Brief Report: IRF4 Newly Identified as a Common Susceptibility Locus for Systemic Sclerosis and Rheumatoid Arthritis in a Cross-Disease Meta-Analysis of Genome-Wide Association Studies. Arthritis Rheumatol. 68, 2338–2344 (2016).
Nakajima, S. et al. Synovial Tissue Heterogeneity in Japanese Patients with Rheumatoid Arthritis Elucidated Using a Cell-Type Deconvolution Approach. Arthritis Rheumatol. 75, 2130–2136 (2023).
Karami, J. et al. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 702, 8–16 (2019).
Dawidowicz, K. et al. The interferon regulatory factor 5 gene confers susceptibility to rheumatoid arthritis and influences its erosive phenotype. Ann. Rheum. Dis. 70, 117–121 (2011).
Duffau, P. et al. Promotion of Inflammatory Arthritis by Interferon Regulatory Factor 5 in a Mouse Model. Arthritis Rheumatol. 67, 3146–3157 (2015).
Cotter, T. G. & Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 158, 1851–1864 (2020).
Patel, S. J. & Liu, N. Hepatic IRF3 fuels dysglycemia in obesity through direct regulation of Ppp2r1b. Sci. Transl. Med. 14, eabh3831 (2022).
Qiao, J. T. et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 81, 13–24 (2018).
Iracheta-Vellve, A. et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 291, 26794–26805 (2016).
He, J. et al. Hepatocyte nuclear factor 1A suppresses innate immune response by inducing degradation of TBK1 to inhibit steatohepatitis. Genes. Dis. 10, 1596–1612 (2023).
Tong, J. et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 69, 2471–2488 (2019).
Li, H. et al. GRP/GRPR enhances alcohol-associated liver injury via the IRF1-mediated Caspase-1 inflammasome and NOX2-dependent ROS pathway. Hepatology 79, 392–408 (2023).
Liang, S. et al. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. J. Biol. Chem. 294, 12359–12369 (2019).
Petrasek, J. et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl Acad. Sci. USA 110, 16544–16549 (2013).
Luther, J. et al. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl Acad. Sci. USA 117, 11667–11673 (2020).
Correction for Luther et al., Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl. Acad. Sci. USA. 117, 16704, (2020).
Du, M. et al. Absence of Interferon Regulatory Factor 1 Protects Against Atherosclerosis in Apolipoprotein E-Deficient Mice. Theranostics 9, 4688–4703 (2019).
Shen, Y. et al. IRF-1 contributes to the pathological phenotype of VSMCs during atherogenesis by increasing CCL19 transcription. Aging (Albany NY) 13, 933–943 (2020).
Fan, X. et al. Non-canonical NF-κB contributes to endothelial pyroptosis and atherogenesis dependent on IRF-1. Transl. Res. 255, 1–13 (2023).
Liu, H. et al. Ablation of Interferon Regulatory Factor 3 Protects Against Atherosclerosis in Apolipoprotein E-Deficient Mice. Hypertension 69, 510–520 (2017).
Seneviratne, A. N. et al. Interferon Regulatory Factor 5 Controls Necrotic Core Formation in Atherosclerotic Lesions by Impairing Efferocytosis. Circulation 136, 1140–1154 (2017).
Edsfeldt, A., Swart, M., Singh, P. & Dib, L. Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture-prone atherosclerotic plaques. Eur. Heart J. 43, 1864–1877 (2022).
Leipner, J. et al. Myeloid cell-specific Irf5 deficiency stabilizes atherosclerotic plaques in Apoe(-/-) mice. Mol. Metab. 53, 101250 (2021).
Tsiantoulas, D. et al. B Cell-Activating Factor Neutralization Aggravates Atherosclerosis. Circulation 138, 2263–2273 (2018).
Senatus, L. et al. RAGE impairs murine diabetic atherosclerosis regression and implicates IRF7 in macrophage inflammation and cholesterol metabolism. Jci. Insight 5, e137289 (2020).
Döring, Y. et al. Hematopoietic interferon regulatory factor 8-deficiency accelerates atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 32, 1613–1623 (2012).
Clément, M. et al. Deletion of IRF8 (Interferon Regulatory Factor 8)-Dependent Dendritic Cells Abrogates Proatherogenic Adaptive Immunity. Circ. Res. 122, 813–820 (2018).
Guo, M. et al. Inhibition of IFN regulatory factor-1 down-regulate Th1 cell function in patients with acute coronary syndrome. J. Clin. Immunol. 30, 241–252 (2010).
Guo, M. et al. IFN Regulatory Factor 1 Mediates Macrophage Pyroptosis Induced by Oxidized Low-Density Lipoprotein in Patients with Acute Coronary Syndrome. Mediators. Inflamm. 2019, 2917128 (2019).
Guo, M. et al. IFN regulatory Factor-1 induced macrophage pyroptosis by modulating m6A modification of circ_0029589 in patients with acute coronary syndrome. Int. Immunopharmacol. 86, 106800 (2020).
Jiang, W., Chen, G. & Pu, J. The transcription factor interferon regulatory factor-1 is an endogenous mediator of myocardial ischemia reperfusion injury. Cell. Biol. Int. 46, 63–72 (2022).
Li, Y. et al. IRF2 contributes to myocardial infarction via regulation of GSDMD induced pyroptosis. Mol. Med. Rep. 25, 40 (2022).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Hu, S. et al. The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int. Immunopharmacol. 107, 108658 (2022).
Liu, Z. et al. Increased sympathetic outflow induced by emotional stress aggravates myocardial ischemia-reperfusion injury via activation of TLR7/MyD88/IRF5 signaling pathway. Inflamm. Res. 72, 901–913 (2023).
Cui, L. et al. Shexiang Tongxin Dropping Pill alleviates M1 macrophage polarization-induced inflammation and endothelial dysfunction to reduce coronary microvascular dysfunction via the Dectin-1/Syk/IRF5 pathway. J. Ethnopharmacol. 316, 116742 (2023).
Courties, G. et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J. Am. Coll. Cardiol. 63, 1556–1566 (2014).
Fan, J. H. et al. Association between IRF-5 polymorphisms and risk of acute coronary syndrome. Dna. Cell. Biol. 29, 19–23 (2010).
Zhang, Y. et al. Interferon regulatory factor 9 is an essential mediator of heart dysfunction and cell death following myocardial ischemia/reperfusion injury. Basic. Res. Cardiol. 109, 434 (2014).
Friesen, M. et al. Activation of IRF1 in Human Adipocytes Leads to Phenotypes Associated with Metabolic Disease. Stem. Cell. Rep. 8, 1164–1173 (2017).
Kumari, M. et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Invest. 126, 2839–2854 (2016).
Yan, S. et al. IRF3 reduces adipose thermogenesis via ISG15-mediated reprogramming of glycolysis. J. Clin. Invest. 131, e144888 (2021).
Mao, Y. et al. STING-IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 37, 920–929 (2017).
Correction to: STING-IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 38, e60 (2018).
Hu, H. Q. et al. The STING-IRF3 pathway is involved in lipotoxic injury of pancreatic β cells in type 2 diabetes. Mol. Cell. Endocrinol. 518, 110890 (2020).
Eguchi, J. et al. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes 62, 3394–3403 (2013).
Cavallari, J. F. et al. Muramyl Dipeptide-Based Postbiotics Mitigate Obesity-Induced Insulin Resistance via IRF4. Cell. Metab. 25, 1063–1074.e1063 (2017).
Sindhu, S. et al. Increased Adipose Tissue Expression of Interferon Regulatory Factor (IRF)-5 in Obesity: Association with Metabolic Inflammation. Cells 8, 1418 (2019).
Sindhu, S. et al. Enhanced Adipose Expression of Interferon Regulatory Factor (IRF)-5 Associates with the Signatures of Metabolic Inflammation in Diabetic Obese Patients. Cells 9, 730 (2020).
Orliaguet, L., Ejlalmanesh, T. & Humbert, A. Early macrophage response to obesity encompasses Interferon Regulatory Factor 5 regulated mitochondrial architecture remodelling. Nat. Commun. 13, 5089 (2022).
Dalmas, E. et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat. Med. 21, 610–618 (2015).
Hoang, A. C., Sasi-Szabó, L. & Pál, T. Mitochondrial RNA stimulates beige adipocyte development in young mice. Nat. Metab. 4, 1684–1696 (2022).
Wang, X. A. et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 305, E485–E495 (2013).
Liu, Z. et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal. Res. 63, e12414 (2017).
Jiang, H. et al. Interferon-α promotes MHC I antigen presentation of islet β cells through STAT1-IRF7 pathway in type 1 diabetes. Immunology 166, 210–221 (2022).
Wang, X. A. et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology 58, 603–616 (2013).
Jiang, D. S. et al. Interferon regulatory factor 1 is required for cardiac remodeling in response to pressure overload. Hypertension 64, 77–86 (2014).
Ma, X. M. et al. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell. Biol. Toxicol. 39, 277–299 (2023).
Yan, M. et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell. Death. Discov. 8, 258 (2022).
Xie, R. et al. LncRNA ZNF593-AS alleviates diabetic cardiomyopathy via suppressing IRF3 signaling pathway. Mol. Ther. Nucleic Acids 32, 689–703 (2023).
Li, N. et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 24, 101215 (2019).
Garcia-Gonzalez, C., Dieterich, C. & Maroli, G. ADAR1 Prevents Autoinflammatory Processes in the Heart Mediated by IRF7. Circ. Res. 131, 580–597 (2022).
Wu, D. et al. Interferon Regulatory Factor-1 Mediates Alveolar Macrophage Pyroptosis During LPS-Induced Acute Lung Injury in Mice. Shock 46, 329–338 (2016).
Liu, S. et al. IRF-1 Intervention in the Classical ROS-Dependent Release of NETs during LPS-Induced Acute Lung Injury in Mice. Inflammation 42, 387–403 (2019).
Chen, X. et al. Interferon regulatory factor 1 (IRF1) inhibits lung endothelial regeneration following inflammation-induced acute lung injury. Clin. Sci. (Lond.). 137, 367–383 (2023).
Wang, N. et al. The STING-IRF3 pathway contributes to paraquat-induced acute lung injury. Toxicol. Mech. Methods 32, 145–157 (2022).
Messaoud-Nacer, Y. et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell. Death. Dis. 13, 269 (2022).
Zhang, S. et al. IFIH1 Contributes to M1 Macrophage Polarization in ARDS. Front. Immunol. 11, 580838 (2020).
Zhang, Y. et al. Anisodamine Enhances Macrophage M2 Polarization through Suppressing G9a-Mediated Interferon Regulatory Factor 4 Silencing to Alleviate Lipopolysaccharide-Induced Acute Lung Injury. J. Pharmacol. Exp. Ther. 381, 247–256 (2022).
Xu, Q. et al. Interferon Regulatory Factor 5 siRNA-Loaded Folate-Modified Cationic Liposomes for Acute Lung Injury Therapy. J. Biomed. Nanotechnol. 17, 466–476 (2021).
Gharib, S. A. et al. Computational identification of key biological modules and transcription factors in acute lung injury. Am. J. Respir. Crit. Care. Med. 173, 653–658 (2006).
Yang, L. et al. Attenuation of interferon regulatory factor 7 activity in local infectious sites of trachea and lung for preventing the development of acute lung injury caused by influenza A virus. Immunology 157, 37–51 (2019).
Winterberg, P. D. et al. Reactive oxygen species and IRF1 stimulate IFNα production by proximal tubules during ischemic AKI. Am. J. Physiol. Ren. Physiol. 305, F164–F172 (2013).
Wang, Y. et al. IRF-1 promotes inflammation early after ischemic acute kidney injury. J. Am. Soc. Nephrol. 20, 1544–1555 (2009).
Yan, Q., Hu, Q. & Li, G. NEAT1 Regulates Calcium Oxalate Crystal-Induced Renal Tubular Oxidative Injury via miR-130/IRF1. Antioxid. Redox Signal. 38, 731–746 (2023).
Yang, X. et al. AhR activation attenuates calcium oxalate nephrocalcinosis by diminishing M1 macrophage polarization and promoting M2 macrophage polarization. Theranostics 10, 12011–12025 (2020).
Liu, H. et al. Erratum: Sulforaphane elicts dual therapeutic effects on Renal Inflammatory Injury and crystal deposition in Calcium Oxalate Nephrocalcinosis: Erratum. Theranostics 12, 421 (2022).
Liu, H. et al. Sulforaphane elicts dual therapeutic effects on Renal Inflammatory Injury and crystal deposition in Calcium Oxalate Nephrocalcinosis. Theranostics 10, 7319–7334 (2020).
Li, Y. et al. IRF-1 promotes renal fibrosis by downregulation of Klotho. Faseb. j. 34, 4415–4429 (2020).
Wu, H., Lai, C. F., Chang-Panesso, M. & Humphreys, B. D. Proximal Tubule Translational Profiling during Kidney Fibrosis Reveals Proinflammatory and Long Noncoding RNA Expression Patterns with Sexual Dimorphism. J. Am. Soc. Nephrol. 31, 23–38 (2020).
Zhao, B. et al. A Genome-Wide Association Study to Identify Single-Nucleotide Polymorphisms for Acute Kidney Injury. Am. J. Respir. Crit. Care. Med. 195, 482–490 (2017).
Renken, I. J. E. et al. No Association between Genetic Loci near IRF2 and TBX1 and Acute Kidney Injury in the Critically Ill. Am. J. Respir. Crit. Care. Med. 201, 109–111 (2020).
Zhang, Y., Zhang, Y., Yang, A. & Xia, F. Downregulation of IRF2 Alleviates Sepsis-Related Acute Kidney Injury in vitro and in vivo. Drug. Des. Devel. Ther. 15, 5123–5132 (2021).
He, T., Yang, L. & Wu, D. Effect of interferon regulatory factor 2 on inflammatory response and oxidative stress in lipopolysaccharide-induced acute kidney injury. Drug. Dev. Res. 83, 940–951 (2022).
Chen, X. et al. Targeting the mechanism of IRF3 in sepsis-associated acute kidney injury via the Hippo pathway. Int. Immunopharmacol. 122, 110625 (2023).
Li, W., Tan, Y., Gao, F. & Xiang, M. Overexpression of TRIM3 protects against LPS-induced acute kidney injury via repressing IRF3 pathway and NLRP3 inflammasome. Int. Urol. Nephrol. 54, 1331–1342 (2022).
Baatarjav, C. & Komada, T. dsDNA-induced AIM2 pyroptosis halts aberrant inflammation during rhabdomyolysis-induced acute kidney injury. Cell. Death. Differ. 29, 2487–2502 (2022).
Xie, J. & Liu, L. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat. Commun. 11, 1600 (2020).
Li, M. et al. Genome-Wide Meta-Analysis Identifies Three Novel Susceptibility Loci and Reveals Ethnic Heterogeneity of Genetic Susceptibility for IgA Nephropathy. J. Am. Soc. Nephrol. 31, 2949–2963 (2020).
Deng, Y. X. et al. Identification and validation of hub genes in drug induced acute kidney injury basing on integrated transcriptomic analysis. Front. Immunol. 14, 1126348 (2023).
Sasaki, K. et al. Deletion of Myeloid Interferon Regulatory Factor 4 (Irf4) in Mouse Model Protects against Kidney Fibrosis after Ischemic Injury by Decreased Macrophage Recruitment and Activation. J. Am. Soc. Nephrol. 32, 1037–1052 (2021).
Chen, M. et al. IRF-4 deficiency reduces inflammation and kidney fibrosis after folic acid-induced acute kidney injury. Int. Immunopharmacol. 100, 108142 (2021).
Gao, Y. et al. Interferon regulatory factor 4 deletion protects against kidney inflammation and fibrosis in deoxycorticosterone acetate/salt hypertension. J. Hypertens. 41, 794–810 (2023).
Lorenz, G. et al. IFN Regulatory Factor 4 Controls Post-ischemic Inflammation and Prevents Chronic Kidney Disease. Front. Immunol. 10, 2162 (2019).
Liu, J., Li, X., Yang, J. & Zhang, D. LncRNA ENSMUST_147219 mediates the progression of ischemic acute kidney injury by targeting the miR-221-5p/IRF6 axis. Apoptosis 27, 531–544 (2022).
Kiryluk, K. & Sanchez-Rodriguez, E. Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy. Nat. Genet. 55, 1091–1105 (2023).
Guo, C. et al. DNA methylation protects against cisplatin-induced kidney injury by regulating specific genes, including interferon regulatory factor 8. Kidney Int. 92, 1194–1205 (2017).
Li, N. et al. IRF8-Dependent Type I Conventional Dendritic Cells (cDC1s) Control Post-Ischemic Inflammation and Mildly Protect Against Post-Ischemic Acute Kidney Injury and Disease. Front. Immunol. 12, 685559 (2021).
Liu, Y. et al. Negative Regulation of SIRT1 by IRF9 Involved in Hyperlipidemia Acute Pancreatitis Associated with Kidney Injury. Dig. Dis. Sci. 66, 1063–1071 (2021).
Bialek, K. et al. Novel association between TGFA, TGFB1, IRF1, PTGS2 and IKBKB single-nucleotide polymorphisms and occurrence, severity and treatment response of major depressive disorder. PeerJ 8, e8676 (2020).
Li, J. et al. Lack of interferon regulatory factor 3 leads to anxiety/depression-like behaviors through disrupting the balance of neuronal excitation and inhibition in mice. Genes. Dis. 10, 1062–1074 (2023).
Li, X. et al. Common variants of IRF3 conferring risk of schizophrenia. J. Psychiatr. Res. 64, 67–73 (2015).
Lago, S. G. et al. Peripheral lymphocyte signaling pathway deficiencies predict treatment response in first-onset drug-naïve schizophrenia. Brain. Behav. Immun. 103, 37–49 (2022).
Xu, Q. et al. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain. Behav. Immun. 81, 495–508 (2019).
Guo, H. et al. ZBP1 mediates the progression of Alzheimer’s disease via pyroptosis by regulating IRF3. Mol. Cell. Biochem 478, 2849–2860 (2023).
Li, R. et al. Silencing of IRF3 alleviates chronic neuropathic pain following chronic constriction injury. Biomed. Pharmacother. 88, 403–408 (2017).
Zhou, Y., Song, W. M., Andhey, P. S. & Swain, A. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020).
Zeng, Q. et al. IRF-8 is Involved in Amyloid-β(1-40) (Aβ(1-40))-induced Microglial Activation: a New Implication in Alzheimer’s Disease. J. Mol. Neurosci. 63, 159–164 (2017).
Huang, Y. et al. IRF1-mediated downregulation of PGC1α contributes to cardiorenal syndrome type 4. Nat. Commun. 11, 4664 (2020).
Wang, A., Kang, X., Wang, J. & Zhang, S. IFIH1/IRF1/STAT1 promotes sepsis associated inflammatory lung injury via activating macrophage M1 polarization. Int. Immunopharmacol. 114, 109478 (2023).
Chen, X. Y. et al. Interferon-regulatory factor-1 boosts bevacizumab cardiotoxicity by the vascular endothelial growth factor A/14-3-3γ axis. Esc. Heart Fail. 11, 986–1000 (2024).
Tanaka, N. et al. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 77, 829–839 (1994).
Testa, U. et al. Impaired myelopoiesis in mice devoid of interferon regulatory factor 1. Leukemia 18, 1864–1871 (2004).
Penninger, J. M. et al. The interferon regulatory transcription factor IRF-1 controls positive and negative selection of CD8+ thymocytes. Immunity 7, 243–254 (1997).
Lohoff, M. et al. Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. Immunity 6, 681–689 (1997).
Hayashi, H. et al. Characterization of dsRNA-induced pancreatitis model reveals the regulatory role of IFN regulatory factor 2 (Irf2) in trypsinogen5 gene transcription. Proc. Natl Acad. Sci. USA 108, 18766–18771 (2011).
Li, M. M. et al. Interferon regulatory factor 2 protects mice from lethal viral neuroinvasion. J. Exp. Med. 213, 2931–2947 (2016).
Lohoff, M. et al. Deficiency in the transcription factor interferon regulatory factor (IRF)-2 leads to severely compromised development of natural killer and T helper type 1 cells. J. Exp. Med. 192, 325–336 (2000).
Tang, P. et al. Regulation of adipogenic differentiation and adipose tissue inflammation by interferon regulatory factor 3. Cell. Death. Differ. 28, 3022–3035 (2021).
Negishi, H. et al. Essential contribution of IRF3 to intestinal homeostasis and microbiota-mediated Tslp gene induction. Proc. Natl Acad. Sci. USA 109, 21016–21021 (2012).
Moore, T. C., Vogel, A. J., Petro, T. M. & Brown, D. M. IRF3 deficiency impacts granzyme B expression and maintenance of memory T cell function in response to viral infection. Microbes Infect. 17, 426–439 (2015).
Santosa, E. K. et al. Control of nutrient uptake by IRF4 orchestrates innate immune memory. Nat. Immunol. 24, 1685–1697 (2023).
Raczkowski, F. et al. The transcription factor Interferon Regulatory Factor 4 is required for the generation of protective effector CD8+ T cells. Proc. Natl Acad. Sci. USA 110, 15019–15024 (2013).
Lien, C. et al. Critical role of IRF-5 in regulation of B-cell differentiation. Proc. Natl Acad. Sci. USA 107, 4664–4668 (2010).
Thomason, H. A. et al. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 120, 1561–1569 (2010).
Wu, M. et al. Interferon regulatory factor 7 (IRF7) represents a link between inflammation and fibrosis in the pathogenesis of systemic sclerosis. Ann. Rheum. Dis. 78, 1583–1591 (2019).
Dror, N. et al. Interferon regulatory factor-8 is indispensable for the expression of promyelocytic leukemia and the formation of nuclear bodies in myeloid cells. J. Biol. Chem. 282, 5633–5640 (2007).
Das, A. et al. Murine IRF8 Mutation Offers New Insight into Osteoclast and Root Resorption. J. Dent. Res. 103, 318–328 (2024).
Wang, P. X. et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J. Hepatol. 62, 111–120 (2015).
Yang, K. et al. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol. Biol. Cell. 17, 1461–1471 (2006).
Probst, P. et al. A small-molecule IRF3 agonist functions as an influenza vaccine adjuvant by modulating the antiviral immune response. Vaccine 35, 1964–1971 (2017).
Hemann, E. A. et al. A Small Molecule RIG-I Agonist Serves as an Adjuvant to Induce Broad Multifaceted Influenza Virus Vaccine Immunity. J. Immunol. 210, 1247–1256 (2023).
Boeszoermenyi, A. et al. A conformation-locking inhibitor of SLC15A4 with TASL proteostatic anti-inflammatory activity. Nat. Commun. 14, 6626 (2023).
Ramírez-Carvajal, L. et al. Expression of porcine fusion protein IRF7/3(5D) efficiently controls foot-and-mouth disease virus replication. J. Virol. 88, 11140–11153 (2014).
Acknowledgements
The study was supported by the Fundamental Research Funds for the National Natural Science Foundation of China (Nos. 82103757, 82273559, 82104373 and 82304063), and Sichuan Natural Science Foundation Project (2023NSFSC1554 and 24NSFSC5087). The authors acknowledge BioRender (https://biorender.com) for the production of some materials in the figures.
Author information
Authors and Affiliations
Contributions
X.J.: Project administration, Supervision, Funding acquisition; X.W.: Conceptualization, Methodology, Supervision; L.W. and Y.Z.: Data curation, Writing-Original Draft, Visualization; N.Z.: Writing-Review & Editing, Visualization; Y.X., Y.T. and J.Y.: Data Curation; G.H.: Visualization; F.R.: Supervision. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Zhu, Y., Zhang, N. et al. The multiple roles of interferon regulatory factor family in health and disease. Sig Transduct Target Ther 9, 282 (2024). https://doi.org/10.1038/s41392-024-01980-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01980-4
This article is cited by
-
Genetic diversity of the immunoglobulin heavy chain locus in cohorts of patients affected with SARS-CoV-2
Human Genomics (2025)
-
Network-based transfer of pan-cancer immunotherapy responses to guide breast cancer prognosis
npj Systems Biology and Applications (2025)
-
Uncovering selection pressures on the IRF gene family in bats’ immune system
Immunogenetics (2025)
-
The rs3757385 polymorphism increases IRF5 expression and systemic nitric oxide metabolites, protecting urothelial bladder cancer patients from recurrence
Molecular Biology Reports (2025)
