
Abstract
Overall response rates in advanced Non-Small Cell Lung Cancer (NSCLC) remains low. Thus, novel molecular targets, tailored drugs and/or drug combinations are needed. Casein Kinase-2 (CK2) is a constitutively active and frequently over-expressed enzyme which fosters tumor survival, proliferation and metastasis. By using a clinical-grade and Cell Penetrating Peptide-based inhibitor coined as CIGB-300, we explore the anti-neoplastic effects caused by interruption of CK2 signaling in lung cancer cells lacking EGFR, ALK and ROS mutations. CIGB-300 penetrated and impaired viability and proliferation of Lung Adenocarcinoma (LUAD) (A549, NCI-H522) and Lung Squamous Carcinoma (LUSC) (NCI-H226 and SK-MES-1) cells in a dose-response manner. The differential activity could not be explained by overall peptide uptake or its subcellular distribution, as evidenced by flow cytometry and confocal microscopy. Upon internalization, CIGB-300 interacted with CK2 catalytic subunits (ɑ1/ɑ2) and CK2 substrates, thus impairing phosphorylation of enzyme substrates (CDC37s13, NPM1s125) and downstream proteins (RPS6s325/326). CK2 inhibition induced an early Reactive Oxygen Species (ROS) and mitochondrial membrane depolarization, which predates lung cancer cell death. Finally, intravenous injection of CIGB-300 in a cell line-based xenograft corroborated CIGB-300’s anti-tumor effects and suggested concurrent in situ reductions of CSNK2ɑ subunit and downstream RPS6s235/236 phosphorylation. Overall, CIGB-300 therapeutic hypothesis and antineoplastic effects demonstrated herein, further support the evaluation of this clinical-grade CK2 inhibitor in advanced NSCLC with limited therapeutic options.
Similar content being viewed by others


Introduction
Lung cancer remains a worldwide problem, independently of improved chemotherapy regimes and significant benefits on selected patient cohorts after targeted or Immune checkpoint therapy1. High-throughput OMICs studies continue providing novel tumor-specific or pan-tumor mutations to intervene; however, the druggability and actual disease-driven potential of such targets remains limited by drug design, tumor heterogeneity and/or drug resistance emergence2,3,4. Albeit limited in efficacy and plagued by unwanted toxicities, chemotherapy still dominates the treatment of advanced non-small cell lung cancer (NSCLC) and new target therapies and/or drug combinations are needed5.
Casein Kinase 2 (CK2) is an emergent oncology target whose over-expression entails a worse patient prognosis6. CK2 activity is usually exacerbated in solid and blood-derived tumors, fostering cell survival, proliferation and metastasis7. The holoenzyme is composed by a variable array of two catalytic (ɑ1/ɑ2) and two regulatory subunits (β), but independent subunits seem to also contribute to malignant phenotype8,9,10. In NSCLC-derived cell lines and tissues, CK2 subunits and the enzymatic activity thereof, were 2-3X higher than their normal counterparts11,12. Particularly, CK2ɑ2 was found over-expressed and associated with distant metastasis both in LUAD and LUSC patients13.
A myriad of CK2 inhibitory compounds have been developed during the last 20 years14. However, thus far only the small ATP-competitive chemical inhibitor CX-4945 and the synthetic peptide inhibitor CIGB-300 have reach clinical grounds15,16. CIGB-300 anti-cancer activities have been demonstrated in pre-clinical models of lung, cervical and breast cancers, as well as T-ALL/CLL/AML leukemias17,18,19,20,21,22. The peptide impairs CK2-mediated phosphorylation by direct binding to a subset of CK2 substrates, but also by direct binding to CK2 catalytic subunits in cancer cells20,23,24. Such dual inhibitory mechanism helps to explain the broad impact of CIGB-300 on tumor cell phosphoproteomes22,23, and also supports the observed direct18, local25 and systemic26 anticancer effects. To note, most of the anticancer effects and mechanistic studies for CIGB-300 in lung cancer models relies on the adenosquamous carcinoma cell line NCI-H12523,26,27, rather a rare and mixed subtype of NSCLC28.
Herein, we focused in the evaluation and characterization of CIGB-300 anti-cancer effects in four cells lines resembling two relevant NSCLC types (i.e. LUAD and LUSC), which also lack actionable oncogenic lesions in EGFR, ALK and ROS4. CIGB-300 antineoplastic effects were re-assessed in vitro and in vivo, in connection with the purported drug therapeutic hypothesis, which includes internalization, CK2 target engagement/inhibition, unleashed subcellular events and ultimate tumor cell demise. Finally, we selected protein/phosphorylation events consistently modulated by CIGB-300 in these experimental models for further evaluation as pharmacodynamic biomarkers on clinical trials for NSCLC with limited therapeutic options.
Materials and methods
Synthetic peptides
CIGB-300 and F20.2 control peptides were synthesized as described before17 on solid phase and purified by RP-HPLC to > 98% purity on acetonitrile/H2O-trifuoroacetic acid gradient and confirmed by ion-spray mass spectrometry (ZPC, Zhejiang, China).
Cell lines
Human NSCLC cell lines A549 (Lung Adenocarcinoma, LUAD)(ATCC: CCL-185, USA), NCI-H226 (Lung Squamous Carcinoma, LUSC)(ATCC: CRL-5826, USA) and NCI-H522 (LUAD)(ATCC: CRL-5810, USA) were cultured in RPMI 1640 medium (Gibco, C11875500CP) and SK-MES-1 (LUSC)(ATCC: HTB-58, USA) cultured in Minimum Essential Medium (MEM) (Gibco, C11095500CP), all supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Gibco, 16000-044) and 1%(v/v) penicillin streptomycin (Gibco,15140-122). Cells were maintained under standard cell culture conditions at 37 °C and 5% CO2.
Crystal violet assay
Antiproliferative effect was determined using crystal violet assay. Cells were seeded in flat-bottom 96-well plates (A549 and NCI-H226, 6 × 103 cells/well; SK-MES-1 and NCI-H522, 8 × 103 cells/well), and 24 h later, serial dilutions of CIGB-300 or selected controls were added and incubated during an additional 48 h. Subsequently, medium was removed and Crystal Violet staining performed (#ab232855; Abcam, Cambridge, UK). Absorbance was measured in a Filter MaxF3 (Molecular Devices, USA), and half-inhibitory concentrations (IC50) values were estimated using CalcuSyn software (v2.1) (Biosoft, Cambridge, UK).
Alamar blue
Cell viability/proliferation was determined using the Alamar Blue assay (Thermo Fisher Scientific, DAL1100). Briefly, cells were seeded as above and 24 h later, serial dilutions of CIGB-300 and CX-4945 drugs were added and incubated for an additional 48 h. Subsequently, Alamar Blue was added as per recommendation from the provider. Cells were further incubated for 5 h at 37 °C, 5% CO2. Fluorescence was measured at 562 nm and 600 nm as a reference wavelength in a FilterMaxF3 (Molecular Devices, USA). The IC50 was estimated after fitting the dose-response curves to the Median-Effect Equation using the software Calcusyn (Biosoft, Cambridge, UK).
Peptide uptake by flow cytometry
Peptide internalization was studied after 3, 10, 30, 45 and 60 min of incubation with 30 μm of a CIGB-300-Fluorecein peptide conjugate (CIGB-300-F; CIGB, Cuba) or with peptide concentrations equivalent to IC50’s for each cell line. Briefly, 6 × 104 or 1 × 105 cells/well were seeded in 24 well plates and incubated for 18–20 h. Subsequently, CIGB-300-F was added in fresh media and incubated during selected time intervals. Following 2 washes, the cells were trypsinized and re-suspended in PBS1X solution. Fluorescent emission was registered in a FACSMelody Flow Cytometer Two laser system (4B-2R configuration) (Becton-Dickinson BD, USA) using a Blue 488 nm laser and D fluorescence detector. A minimum of 20,000 events were acquired for each experimental sample and the data analysis was performed using the FlowJo software v10.
Peptide uptake by confocal microscopy
Fluorescence microscopy experiments were performed on a confocal microscope (CFM) (Leica TCS SP8, Wetzlar, Germany) equipped using 488 nm laser. Briefly, cell suspensions of 1 × 105 cells/ml were cultivated for 20–24 h in suitable cell culture chambers at 37℃ and 5% CO2. Next day, CIGB-300-F conjugates were added to cell cultures at final concentrations of 30µM or IC50 and incubated for 10, 30, and 180 min. Afterwards, cells were stained for 15 min with 2 ug/ml Hoechst 33,342 solution (Sparkjade, CU0002) and subsequently washed with PBS1X. Finally, fixed cells were immediately analyzed by CFM with a stage top incubator (Tokai Hit, Shizuoka, Japan). Image analysis was performed using LAS X software (Leica, Wetzlar, Germany) and Image J software (National Institutes of Health, Bethesda, MD, USA).
In vivo pull-down
Pull-down experiments were performed as described17. Briefly, 3 × 106 cells were seeded in T75 flasks (8 ml) and incubated during 20 h in cell culture conditions. Next day, cells were incubated for 45 min with a concentration of CIGB-300 conjugated to biotin (CIGB-300-B) corresponding to IC90 for each cell line. Afterward, tumor cells were lysed by freeze-thaw cycles (8X; liquid nitrogen/37 °C) in an hypotonic PBS solution (PBS0.1X) containing 1 mmol/L DTT (Sigma, MO, USA), 0.1% Triton™ X-100 (Sigma, MO, USA) and complete protease inhibitor (Thermo, Rockford, USA). Then, the cellular lysate was cleared by centrifugation during 15 min at 12 × 103 rpm/4ºC and the protein contained in the total protein content in the collected supernatant quantified. Finally, 200 µg of total protein were added to 30 µL of pre-equilibrated streptavidin-sepharose matrix (GE Healthcare Uppsala, Sweden) and incubated during 1 h/4ºC. CIGB-300´s-interacting proteins were subsequently eluted, separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes (BioTrace NT nitrocellulose#66485, Pall Life Sciences, USA) for Western Blot (WB) using a Trans-Blot® Turbo™ Transfer System (BIORAD, USA).
Western blot-based signaling studies
Total cell lysates (TCE) for signaling experiments were prepared as described29, and equal amounts of protein (30 µg/sample) were resolved by 12% SDS-PAGE, transferred as above and immunoblotted with the following antibodies: p-AKT (S129)(s473)/AKT, p-PTEN (S380)/PTEN, p-RPS6 (S235/236)/RPS6 and PARP1, all from Cell Signaling Technology (USA). In addition, B-actin (Sigma, USA); p-B23/NPM1 (S125)/NPM1 (Abcam, Cambridge, UK) and CK2α1/α2 (Abcam, Cambridge, UK) antibodies were also used. After immunoblotting, detection was performed by incubation with horseradish peroxidase-conjugated anti–mouse, anti–rabbit (Sigma, USA), or anti–goat (Santa Cruz Biotechnology) immunoglobulin (IgG; 1:5000 dilution), Detection was carried out with SuperSignal™ West Dura Extended Duration Substrate reagent (Thermo Fisher Scientific#34076). Images and subsequent densitometry analysis were performed using Ibright1500 device and software (Thermo Fisher, USA). Integrated density values for each band of interest were normalized to loading controls in each lane/condition.
ROS detection
For detection of reactive oxygen species (ROS) levels, the cells were incubated with selected peptide concentrations (IC50) during 1–6 h. Then, 3 µM of the fluorescent probe dihydroethidium (DHE) (Sigma, MO, United States) was added and incubated during 30 min at 37 °C in the dark. Then, cells were washed with PBS1X and prepared for analysis by CFM (Ex-488 nm) or Flow cytometry (Ex-488 nm, Detector C). In some experiments, 20 mM N-acetyl cysteine (NAC, Sigma), F20.2, or CX-4945 were used as controls at indicated concentrations and time incubation.
Mitochondrial membrane potential
The JC-1 Mitochondrial Membrane Potential Detection Kit (Invitrogen, M34152) was used to estimate changes in mitochondrial membrane potential by CFM. JC-1 dual-fluorescence emission was measured after incubating tumor cells with selected peptide concentrations (IC50) during 1–6 h. Following incubation, cells were stained with JC-1 for 20 min at 37℃, and then washed with PBS 1X. Finally, cells were analyzed by CFM with green (488 nm) and red (638 nm) lasers. Change of mitochondrial potential were shown by estimation of red/green fluorescence intensity ratio. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), F20.2, CX4945 were used as control where indicated.
Quantitative real-time PCR
Tumor cells were incubated with CIGB-300 concentrations corresponding to IC50 for each cell line or vehicle during 0.5 h, 3–6 h and then total RNA isolated and quantified (1 × 106 cells/condition). In some instances, cells treated with CX4945 (5 µM, 6 h) or corresponding vehicle (DMSO), were also processed. Briefly, after incubation cells were washed with PBS1X and suspended in 350 µL of RLT lysis buffer with 1% β-mercaptoethanol (Sigma, St. Louis, MO, USA) for total RNA isolation (RNeasy Plus Kit, Qiagen). Total RNA samples were checked by Nanodrop spectrophotometer to measure quality control parameters [concentration (ng/µL) and OD relation (1.8 to 2.2 units, 260/280nm ratio)]. Complementary (c)DNAs were obtained from 1ug of total RNAs, using the Transcriptor First Strand cDNA Synthesis Kit package (Roche, Mannheim, Germany). The qPCR reactions were set up in 20 µL using LightCycler® 480 SYBR Green I Master 2X (Roche, Mannheim, Germany), 300 nM of oligonucleotides, and three replicates per sample of 1:10 cDNA dilution (Table S1). Runs were carried out in the LightCycler® 480II equipment (Roche, Mannheim, Germany) in a 96-well format and SYBR Green Probe II mode with a standard program with 45 cycles. Gene expression changes for 45SrRNA and NPM1/B23 transcripts were expressed as a ratio in relation to untreated time-matched control using Cp and efficiency for the calculations in REST 2009 program, after normalization with ACTB and DDX5 genes30. This program reports a p-value after a pair wise fixed reallocation randomization test, reporting increasing and decreasing gene levels; here we report those changes for a p < 0.05. Two independent experiments with three replicates per condition were performed.
Apoptosis detection by flow cytometry
Viability of lung cancer cells incubated with CIGB-300 was assessed using the FITC AnnexinV Apoptosis Detection Kit I (BD Bioscience, USA). Briefly, cells were incubated with equipotent concentrations of CIGB-300 (IC50), unless state otherwise, during 0.5, 1, 3, 6–24 h; then washed twice with cold PBS1X and suspended in binding buffer (1X) at 1 × 106 cells/ml. Next, 5 µl of FITC AnnexinV and 5 µl of Propidium Iodide (PI) were added and incubated in dark for an additional 15 min. Finally, cells were washed with cold PBS1X and subsequently analyzed by Flow Cytometry with the acquisition of 20,000 events/sample in the instrument described above.
Animal experiments
Male and Female SPF-grade BALB/c mice (18–22 g) were purchased from Hunan STA Laboratory Animal Co., LTD. with the license No. of SCXK (Xiang) 2019-0004, and maintained in Area D of Hunan Prima Pharmaceutical Research Center Co., LTD. with the license No. of SYXK (Xiang) 2020-0015. All procedures were operated according to the Guide for the Care and Use of Laboratory Animals (Eighth Edition) issued by The National Academics (Washington, DC). The study was approved by the Animal Ethics Committee of Hunan Provincial Research Center for Safety Evaluation of Drugs. The study was performed according to the ARRIVE Guidelines (https://arriveguidelines.org/). NCI-H226 cells (1 × 107/mL) were inoculated subcutaneously into 24 BALB/c-nu nude mice at the right armpit (0.2 mL/mouse). Once average tumor size reaches 30 mm3, 18 tumor-xenografted mice with no ulceration were randomized into the following treatment groups (6 mice/group): Vehicle, CIGB-300 (2 mg/kg), and CIGB-300 (10 mg/kg) (3 females + 3 males each). Each group received two cycles of treatments (each cycle composed by five consecutive injections, 1 per day), with 6 days of drug-holidays in between. After last administration, 2 animals/group were euthanized using inhaled isoflurane, and cervical dislocation was used as secondary euthanasia before to collect tumors for IHC analysis. The rest of mice were kept to record tumor volume and survival. Tumors were measured with a caliper and volumes calculated using the formula: tumor volume (TV) = 1/2 x a (length) x b2 (width).
Statistical analysis
Unpaired t-test (two-tailed) were performed to compare selected experimental groups on ROS (DHE) and mitochondria depolarization (JC-1) experiments (GraphPad Prism v6.01, CA, USA). For animal data, the SPSS23.0 statistical software was used. Measured data was expressed as mean ± standard deviation, and Leven’s test was used to test normality and homogeneity of variance. If there is no statistical significance (P > 0.05), one-way analysis of variance (ANOVA) was performed. If ANOVA is statistically significant (P ≤ 0.05), the Least Significant Difference (LSD) test (parametric method) was used for comparative analysis. If variances are unequal (P ≤ 0.05), a Kruskal-Wallis followed by Dunnett’s Test (non-parametric method) was conducted for experimental groups comparison. Survival analysis was performed by Longrank test.
Results
CIGB-300 impairs the viability and proliferation of NSCLC lacking actionable targets
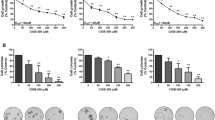
To demonstrate that CIGB-300 impairs the viability and proliferation of NSCLC cells representing two major types, we conducted in vitro experiments in two lung adenocarcinoma (LUAD: A549 and NCI-H522) and two lung squamous (LUSC: NCI-H226 and SK-MES-1) cell lines. A concentration-dependent cytotoxic effect for CIGB-300 was observed within the range of 35–150 µM, as assessed by Alamar Blue (AB) and Crystal Violet (CV) assays (Fig. 1a, b).

Concentrations-dependent effects of CIGB-300 over viability and proliferation of Lung Adenocarcinoma (LUAD) or Lung Squamous Carcinoma (LUSC) cells as evaluated by Alamar Blue (a) or Crystal Violet (b) assays after 48 h of incubation. The experimental points were fitted to a log(agonist) vs. response -- Variable slope (four parameters) model to construct the observed dose-response curves. Fraction Affected (FA) refers to the cytotoxic effect exerted by the drug at a given concentration according to the formula FA=(1- abs[drug]/abs[veh]). The estimated IC50 values are shown in the graph. Confidence intervals and curve fitting parameters are available at Supplementary Table S2 online. The negative control peptide (F20-2) comprise the same CPP (Tat), but not the inhibitory cargo (p15).
Of note, in three of the four cell lines, the IC50’s values determined by CV assay were almost half of those estimated by AB assay (Fig. 1; Supplementary Table S2 online). Differences on IC50 values according to cellular readouts, were also observed for CX-4945, a small chemical compound inhibitor of CK2 (Supplementary Fig. S1; Supplementary Table S3 online). Overall, CIGB-300’s potency was higher in LUAD than LUSC cells, whereas not such a trend was observed for CX-4945 (Fig. 1; Supplementary Fig. S1 online; Supplementary Table S2, S3 online).
CIGB-300 differentially penetrates NSCLC of LUAD and LUSC origin
Cell penetration and productive subcellular distribution are major barriers for therapeutic effects of CPP-based drugs like CIGB-300. Thus, the observed cytotoxic and antiproliferative effects could be explained by differences on peptide uptake across target cells. To address this, we analyzed peptide internalization on live cancer cells by flow cytometry and Confocal Microscopy in concentration-dependent and time-course experiments (Figs. 2 and 3).

Short-term internalization of CIGB-300 peptide in live NSCLC cells as analyzed by flow cytometry. (a) Representative FSC vs. SSC dot-plots (upper panel) and FL1-Histograms (lower panel) obtained after incubation of A549 cells with 30 μm or an equipotent concentration (IC50) of a peptide-fluorescein conjugate during 3, 10, 30, 45 and 60 min. Live cells on FSC vs. SSC plots were selected to estimate the Geometric mean of the fluorescence on FL1 channel in arbitrary units (GmeanF, a.u.). Vehicle-treated cells were used to set an auto-florescence threshold to score per cent of cells displaying florescence (%CIGB-300-F+). (b) Estimation of %CIGB-300-F + cells (upper-panel) and magnitude of peptide internalization (GmeanF, a.u., lower-panel) for the four cell lines in study (n = 3). (c) The collected GmeanF values (a.u) were fitted to a first-order exponential association model to obtain the kinetic parameters and confidence intervals described in the Table; Ymax, maximum Fluorescence at the curve plateau; K, rate constant of internalization; Half-time = ln(2)/K; R square, Goodness of fit.
By using a fixed peptide concentration (30 μm) and incubation time (0–1 h), which are clinically relevant since the pharmacokinetics point of view, we verified a fast uptake of CIGB-300 in the four cell lines (Fig. 2a, b). As early as 3 min after incubation, 40–70% of the cell populations displayed intracellular florescence (i.e. CIGB-300-F+). These values reached up to 95% of whole cell populations when peptide concentrations representing the IC50 were used for each cell line (Fig. 2a, data not shown). Similar uptake levels where attained at 30 μm of concentration, but after 30 min of incubation with the peptide. To note, 30 μm of CIGB-300 caused no impact on cell viability (i.e. AB readout), but exerted 18 and 33% of proliferation inhibition (i.e. CV readout) on LUAD cells A549 and NCI-H522, respectively (Fig. 1a, b). Altogether, a fast and concentration-dependent CIGB-300 penetration in tumor cell lines were observed.
On the other hand, internalization curves and estimated parameters indicate that a steady-state peptide uptake was reached around 45 min of incubation with 30 μm of peptide (Fig. 2b, c). At such fixed concentration, the speed of internalization (i.e. rate constant “K”) tended to be higher in LUAD than LUSC cells, although data dispersion (i.e. R square, 95% CI) precludes any conclusion. Overall, the magnitude of internalized CIGB-300 was higher in NCI-H226 (LUSC) than in the rest of the cell lines, at two tested peptide concentrations (Ymax, 30 μm = 370.8 a.u.; Ymax, IC50 = 1027 a.u.) and according to short-(< 1 h) and long-term (0–24 h) uptake experiments (Fig. 2c; Supplementary Fig. S2 online). At equipotent concentrations (IC50) peptide internalization tends to be higher in LUSC cells than in LUAD cells, whereas the steady-state was reached after 45–60 min of incubation (Supplementary Fig. S2 online). Finally, higher peptide concentrations (IC50’s) produced peaks internalization values, albeit such dose-dependent uptake was not linear (Fig. 2c vs. Supplementary Fig. S2 online).
We have demonstrated that CIGB-300 capacity to reach the nuclear compartment correlates with its antiproliferative activity31. Therefore, we analyze CIGB-300 subcellular distribution by using live-cell microscopy in concentration- and time-course experiments. As expected, longer incubation times and higher peptide concentrations, leads to increased intracellular peptide in both LUAD and LUSC cells (Supplementary Fig. S3 online). In line with flow cytometry data, almost whole cell populations became positive for peptide uptake when incubate with IC50’s values; however, LUSC cells showed higher peptide uptakes than A549 (LUAD) even when equipotent CIGB-300’s concentrations were used (Fig. 3a, b; Supplementary Fig. S3a, b online).

Representative CIGB-300’s internalization studies performed by live-cell CFM at indicated time points. (a) NCI-H226 cells (b) A549 cells were incubated during 30 and 180 min using equipotent concentrations (IC50’s) of fluorescein-conjugated CIGB-300 and subsequently analyzed by CFM. ( c, d) The percent of cells displaying nucleolar fluorescence (nucleoli+%) was scored at two peptide concentrations 30 μm (c) and IC50’s (d). Hoeschst 33342 reagent was used to visualize the cell nucleus. The magnification was settled as optical zoom 20X + 1X digital zoom. A selected zoom in the Region of Interest (ROI) was further digitally magnified 2X (a, b-right panel). BF, Bright Field.
To note, peptide internalization comprised a variable pattern of diffuse or punctuated cytoplasmic distribution across inspected Z levels at CFM, without gross differences between LUAD and LUSC cells (Fig. 3a, b and 2× magnification). However, there was a different concentration- and time-dependent nucleolar deposition across the cell panel (Fig. 3c, d). Particularly, at equipotent concentrations, only A549 and SK-MES-1 cells showed a percent value of nucleoli + cells close or above the 50% of the cell populations (Fig. 3d), yet such nucleolar deposition only induced 10–12% of nucleolar disassembly after 3 h of peptide incubation according to bright field CFM analysis (Supplementary Fig. S4a, b online). On the contrary, less than 20% of NCI-H522 cells displayed nucleolar staining in spite of using CIGB-300’s concentrations causing 50% and 75% of viability and proliferation impairment, respectively (Fig. 1a, b).
Overall, higher IC50’s values in LUSC cells were associated with a higher CIGB-300 uptake (Supplementary Table S2; Supplementary Fig. S2 online), but not necessarily with an increase on its nucleolar deposition and/or subsequent nucleolar disassembly activity as measured by CFM (Fig. 3d; Supplementary Fig. S4a, b online). However, at molecular level, when 45SrRNA and 45SrRNA/NPM1 ratios were estimated by qPCR, we observed a more consistent decreasing of both parameters in LUSC than in LUAD cells after 3 h of peptide incubation (Supplementary Fig. S4c, d online). To note, the CK2 inhibitor CX-4945 did not significantly decreased 45SrRNA levels in tested cells.
CIGB-300 impairs CK2-mediated signaling in both NSCLC types
Previously, we demonstrated that CIGB-300 binds to CK2 catalytic subunits and nucleolar protein B23/NPM1 in an adenosquamous cell line23. To verify whether such interactions also account in LUAD and LUSC, we performed in vivo pull-down followed by western blot experiments in A549 and NCI-H226 cells. Figure 4a, b shows that CIGB-300 interacts with all CK2 subunits i.e. CK2α1, CK2α2 and CK2β, as well as with the CK2 substrate NPM1, but not with AKT in both A549 and NCI-H226 cells.

In vivo CIGB-300 interactions with selected intracellular proteins and CK2-mediated signaling inhibition on A549 and NCI-H226 cells. (a, b) Pull-Down (PD) coupled to WB was performed to verify peptide interactions with CK2 subunits and selected substrates in LUAD (A549) and LUSC (NCI-H226) cells. CIGB-300-B conjugate at a concentration corresponding to its IC90 was incubated during 45 min with target cells and subsequently submitted to PD. (c) WB of TCE from A549 cells incubated with CIGB-300 (IC50) from 1–6 h, as well as relevant controls. TCE, Total Cell Extract; In+, Input for PD from cells incubated with CIGB-300(+); In-, Input for PD from cells incubated with vehicle (-); Pass, non-bound fraction or PD supernatant; PD+, CIGB-300 interactions; PD-,vehicle interactions; veh, cells incubated with vehicle; +, cells incubated with CIGB-300; CX, cells incubated with CX-4945 (IC50); F20-2, cell incubated with negative control peptide at the same concentration as CIGB-300. Blots strips in (a) and (b) were cropped from different parts of the same membrane as indicated by white in-between lines. Blots strips in (c) were cropped from different membranes as indicated by the inclusion of B-actin I (Gel 1) or B-actin II (Gel 2) at the bottom. Numbers shown in (c) were estimated after densitometric image analysis (ImageJ) as the ratio of each protein band relative to B-actin (total proteins) or relative to corresponding total proteins (phosphoproteins). Images are representative of two independent experiments. Raw images can be found at Supplementary Information File#2.
Subsequently, we performed WB with Total Cell Extracts (TCE) from CIGB-300’s and vehicle incubated cells to corroborate that such CIGB-300-CK2 interactions impair CK2 enzymatic activity. As expected, incubation with equipotent concentrations of CIGB-300 inhibited total/phosphorylation levels of direct substrates like CDC37/CDC37s13 and NPM1/NPM1s125, as well as downstream phosphorylation events linked to CK2 (RPS6s235/236), both in LUAD (A549) and LUSC (NCI-H226/SK-MES-1) cells (Fig. 4c; Supplementary Fig. S5 online). To note, while such inhibition seemed transient in NCI-H226 cells (3 h), it further deepens in A549 and SK-MES-1 cells (6 h). Concerning the NPM1/NPM1s125 protein, a slight decrease was observed after 6 h of incubation in A549 LUAD cells, in a contrast with the inhibition observed in NCI-H226 and SK-MES-1 cells (Fig. 4c; Supplementary Fig. S5a, b online).
The above described inhibitory effect of phosphorylation could be explained by a decrease in CK2 catalytic subunits upon incubation with CIGB-300. Protein levels of CK2α1 in A549 (LUAD) and CK2α2 in NCI-H226 and SK-MES-1 (LUSC) decreased when cells were incubated with equipotent concentrations of CIGB-300; whereas, the effect of CIGB-300 on CK2β regulatory subunit levels seems negligible in these cell lines (Fig. 4c; Supplementary Fig. S5a, b). Otherwise, in NCI-H522 cells we observed clear reductions for all the three CK2 subunits upon CIGB-300 incubation, as well as a consistent decreasing on CDC37s13 substrates phosphorylation (Supplementary Fig. S5c online). Finally, as expected, incubation with the CK2 inhibitor CX-4945 inhibited downstream RPS6s235/236 phosphorylation in tested cell lines; whereas, also impaired CDC37s13 phosphorylation in SK-MES-1 and NCI-H522 cells (Supplementary Fig. S5b, c online).
CIGB-300 induces mitochondrial depolarization and ROS production
Previous findings indicate CIGB-300 induce mitochondrial membrane depolarization and Reactive Oxygen Species (ROS) in cancer cells29,32. Having demonstrated that CIGB-300 impairs CK2-mediated signaling in LUAD and LUSC cells, we evaluate the impact of such inhibition on mitochondrial potential by measuring JC1 fluorescence emission in time-course experiments (Fig. 5).

CFM images showing JC1-Red/Green fluorescence ratio as marker for mitochondrial membrane depolarization induced by CIGB-300. (a) Representative images after 6 h incubation with CIGB-300 (IC50) in A549 cells; The quantification of four independent fields (> 10 ROI) is shown for A549 (b) and NCI-H226 (c) cells. CCCP, positive control. veh, Vehicle incubated cells. Images were acquired with an optical zoom of 20X and digital zoom of 1X. Selected groups were compared against veh using an Unpaired t-test (two-tailed) and statistical differences, when found, denoted as: *, p < 0.05; **,p < 0.01; ***, p < 0.001.
CIGB-300 induced a time-dependent mitochondrial membrane depolarization which was earlier and deeper in A549 than NCI-H226 cells, and more noticeable after 6 h of incubation. To note, CX-4945 showed a similar pattern at tested conditions, with a clear depolarization effect on A549 LUAD, but no effect in NCI-H226 LUSC cells after 6 h of incubation (Fig. 5b, c).
Mitochondria impairment has been linked to unbalance ROS production; thus, we explore whether CIGB-300 induce ROS by flow cytometry and CFM experiments (Fig. 6; Supplementary Fig. S6, S7 online).

ROS detection by flow cytometry after incubation of LUAD cells with CIGB-300 as revealed by DHE method. (a) Representative analysis with FSC vs. SSC and histograms plots (Overlays) corresponding to A549 cells incubated during 6 h with CIGB-300 at IC50 concentration value; In the overlay histograms plots a DHE high population emerged on CIGB-300 incubated cells, with mean Fluorescence values (GMeanF) in arbitrary units (a.u) showed as inserts. (b) Quantification of GmeanF on A549 cells treated as (a); (c) Quantification of GmeanF on NCI-H522 cells incubated during 6 h with IC50 concentration. Veh, vehicle; NAC, N-acetyl cysteine. Selected groups were compared against Veh using an Unpaired t-test (two-tailed) and statistical differences, when found, denoted as: *, p < 0.05; **,p < 0.01; ***, p < 0.001. Square ** refers to statistical analysis for ROS quenching effects from NAC, i.e. CIGB-300 vs. CIGB-300 + NAC groups; H2O2 vs. H2O2 + NAC.
CIGB-300 induced significant ROS levels after 6 h incubation in both LUAD and LUSC cells, as evidenced by flow cytometry (Fig. 6; Supplementary Fig. S7 online) and CFM (Supplementary Fig. S6 online). To note, no major differences between LUAD and LUSC cells concerning the magnitude of ROS induction by CIGB-300 could be found; however, in correspondence with mitochondrial depolarization, ROS induction was only significant after 6 h of peptide incubation (Supplementary Fig. S6 online) and could be registered up to 24 h (Supplementary Fig. S7, S8 online).
ROS induction by CIGB-300 was associated with an upward-left shift of a fraction of cells out of “live cells” gate in SSCvsFSC plots (Fig. 6a insert; Supplementary Fig. S8 online). Such “emerging” population displayed higher intracellular ROS according to DHE emission. As expected, NAC reduced DHE fluorescence emission on CIGB-300 treated cells, but at the same time counteracted the cytotoxicity induced by the peptide, as reflecting by around 50% reduction on the SSC-high/FSC-Low cell populations in A549 and NCI-H522 cell lines (Fig. 6a; Supplementary Fig. S8 online). Unfortunately, this SSC-high/FSC-Low approach could not be followed in LUSC cells; thus, a putative NAC protective effect in NCI-H226 or SK-MES-1 cells could not be measured (Supplementary Fig. S7, S9 online).
CIGB-300 induces cell death irrespective of NSCLC type
To verify whether these CIGB-300’s induced molecular and subcellular events triggered cell death by apoptosis, we measured time- and concentration-dependent exposition of phosphatidylserine on peptide incubated cells. Indeed, CIGB-300 induced cell death in both NCSLC types, in a time-course and dose-response manner (Fig. 7a, b; Supplementary Fig. S9 online). Of note, at IC90 equivalent peptide concentration around 1/3 of cell population showed early (3 h) signals of phosphatidylserine exposition (Fig. 7a); however, cell death tends to peak at 24 h and was mainly characterized by a double positive cell staining (i.e. AnnexinV+/PI+) (Fig. 7a, b; Supplementary Fig. S9 online). In A549, cell death was also evidenced by a decrease in the so-called “alive cell population” gate in FSC vs. SSC flow cytometry dot-plots (Fig. 7a, bottom panel).

Cellular and molecular markers of apoptotic cell death on NSCLC cells incubated with CIGB-300 in concentration-dependent and time-course experiments. (a) Representative flow cytometry dot plots, upper panel: X-axis AnnexinV-Fluorescein(FITC) vs. Y-axis (PI) and lower panel: X-axis FSC vs. Y-axis SSC, obtained after AnnexinV-PI staining of A549 cells incubated with CIGB-300 (IC90) during indicated time points. Percent of cell populations is indicated within each quadrant, for Q2 mean ± SD of three replicates; veh, vehicle. (b) Summary of the AnnexinV-PI staining experiments on the four cells lines, at a fixed time point of 24 h using two peptide concentrations (IC50 & IC90). Here, AnnexinV + comprised both A+/PI + and A+/PI- cell populations. (c) WB for detection of PARP cleavage after incubation of A549 or SK-MES-1 cells with CIGB-300 (IC50) during 3–6 h. B-actin protein was used to normalize total protein content/lane; +, CIGB-300; CX, CX-4945; veh, vehicle incubated cells, for CIGB-300 (PBS1X) and for CX-4945 (DMSO). Molecular weights of full length PARP (118 kDa), expected PARP cleavage product (89 kDa) and B-actin (43 kDa). Blots strips in (c) were cropped from different parts of the same membrane as indicated by white in-between lines. Images are representative of two independent experiments. Raw images can be found at Supplementary Information File#2.
To further investigate about CIGB-300’s induced cell death type, we measured by WB PARP and Caspase-3 cleavage in TCE from A549, SK-MES-1 and NCI-H226 cells incubated with the peptide. CIGB-300 peptide induced an early (3–6 h) full length PARP cleavage (i.e., reduction of 116 kDa band); however, this was not paralleled by an increase in the 89 kDa cleavage product, neither after short-time incubation (A549, SK-MES-1) nor after longer incubation times (A549, NCI-H226) (Fig. 7c; Supplementary Fig. S10a online). Otherwise, WB for Caspase-3 detection in A549 and NCI-H226 cells also evidenced a decreasing of full-length Caspase-3 protein upon incubation with CIGB-300, but did not shown the expected cleavage products either (Supplementary Fig. S10b online).
CIGB-300 reduces LUSC tumor growth and modulates CK2 signaling in vivo
To evidence that CIGB-300’s effects over viability/proliferation of LUSC cells translate into anti-tumor activity in vivo, we recorded tumor growth delay (TGD) following intravenous injections of CIGB-300 in a NCI-H226 cell-line derived xenograft. Two cycles of 5-consecutive peptide injections (2 or 10 mg/kg) induced a significant TGD at day 16 post-administration (Fig. 8a). No differences on TGD were seen between these two peptide doses; however, the survival data showed that mice treated with 10 mg/kg survived 16 and 12 days more than vehicle- and 2 mg/kg-treated mice, respectively (Fig. 8b).

Anti-tumor effect of CIGB-300 administered by intravenous injection in a cell line-based NCI-H226 xenograft tumor model. Two cycles of 5-consecutive injections of CIGB-300 at 2 mg/kg or 10 mg/kg were injected by the tail vein and Tumor Growth (a) or Survival (b) scored until day 16 or day 40, respectively. At day 16, two mice per group were sacrificed for IHC analysis on tumor mass using indicated antibodies (c), whereas the rest were maintained for survival analysis (n = 4). In (a), ∆ indicate peptide injections. At every time-point, Tumor Volume (means ± SD) of CIGB-300-treated mice were compared against Vehicle-treated mice by one-way ANOVA followed by Dunnett multiple comparison test and denoted when significant ( * p < 0.05, * * p < 0.01). For survival analysis (b), Kaplan-Meier Curves were compared by a Logrank (p = 0.0022) and Logrank test for trend (p = 0.0005), as well as by individual pair-wise comparison against Vehicle-treated mice and denoted when significant (** p < 0.01). Median survival in days (d) for each experimental group is also shown. In (c), representative IHC pictures and staining scores are shown as insert (mean ± SD). No statistical analysis was done since only 2 mice per group were analyzed here (Supplementary Table S3 online).
To preliminary explore the putative impact of CIGB-300 intravenous injections on CK2 signaling in tumor mass, we conducted IHC analysis on tumor samples taken from two of the six vehicle- or CIGB-300-treated mice from each group. Our results indicated that CIGB-300 tend to decrease in around 50% the CK2 catalytic subunit CK2ɑ1 and downstream phosphorylation event RPS6s235/236, with an apparent dose-response pattern for these two proteins (Fig. 8c; Supplementary Table S4 online). However, the effects of these peptide doses and administration regimes on CK2ɑ2 and CDC37s13 direct enzyme substrate were less evident and shall be further explored.
Discussion
CK2 is an emerging oncology target which clinical validation is ongoing by using divergent inhibition strategies15,16. CIGB-300 herein, is a chimeric synthetic peptide composed by a CPP (Tat) fused to an inhibitory payload (p15) aimed to block CK2 intracellular signaling by direct binding to oncogenic substrates17. However, recent evidences support a dual-inhibitory mechanism for CIGB-300, which also interacts with CK2 catalytic subunits and/or CK2 holoenzyme in a yet to be disclosed site23. To note, most of CIGB-300 research on pre-clinical lung cancer models have been conducted in an adenosquamous carcinoma cell line23,26,31, a rare and mixed subtype of NSCLC28.
Here, we further investigate into the mechanism of action of CIGB-300 by using LUAD and LUSC cell models lacking major NSCLC actionable mutations1. EGFR-, ALK- and ROS1-mutated NSCLCs are so-called ‘oncogene-addicted’ tumors, which can be treated using 3 to 5 different FDA-approved targeted drugs4. Such mutations are absent from our selected cell lines according to their previous comprehensive characterization https://cancer.sanger.ac.uk/cell_lines33 and https://depmap.org/portal/34. In addition, we evaluate phosphorylation events consistently down-regulated by CIGB-300 in such cellular models in order to identify putative pharmacodynamic biomarkers for this particular CK2 inhibitor.
Peptide internalization is an important barrier for a CPP-based drug35,36. Our results indicate that CIGB-300 penetrates virtually 100% of cells populations within 3–30 min, with half-life times from 7 to 17 min depending on the peptide concentration and the cell line tested. However, at equipotent concentrations (IC50) and short-term incubations (0–60 min), LUSC cells showed higher intracellular peptide accumulation than LUAD. This trend was more evident when peptide incubation was extended up to 24 h. While these findings can not be generalized, may indicate that CIGB-300’s internalization in LUSC cells could be “less productive” since more intracellular peptide is needed to achieve an equivalent cytotoxic effect. Peptide internalization, vesicular transportation and intracellular degradation are variables linked to CIGB-300’s in vitro antineoplastic effects27. A “productive internalization” for a CPP-based drug like CIGB-300 comprise all the way along since membrane penetration, intracellular traffic/degradation until target(s) engagement and inhibition in a given subcellular compartment.
Studies involving lung, cervical, prostate and colon cancer cells, evidenced that CIGB-300’s antiproliferative effects correlated with its nucleolar deposition31. In line, one of the first targets identified for CIGB-300 was the nucleolar protein NPM137. Herein, our Confocal Microscopy results corroborate time-course and dose-response penetration of CIGB-300 into LUAD and LUSC cells. However, the intracellular distribution of CIGB-300 was variable, and reaching/disassembling the nucleoli seems no critical for its cytotoxic effect, as exemplified by NCI-H522 and SK-MES-1 cells, respectively. The observed mixture of punctuate and diffuse staining suggest the co-occurrence of several internalization pathways as described before for arginine-rich CPP38 and for CIGB-30027. To note, in spite of lower-than-expected nucleolar deposition for CIGB-300 in 3 of 4 cell lines tested, we noticed that peptide concentration, gross uptake and nucleolar impairment (45SrRNA-qPCR), are somehow related as expected for a Tat(CPP)-based drug39.
Following peptide internalization, CIGB-300 interacted with CK2ɑ1, CK2ɑ2 and CK2β regulatory subunits. However, former cell free-experiments demonstrated that CIGB-300 interacts with CK2ɑ and not CK2β; thus, raising the possibility that CK2β herein was identified as part of the holoenzyme complex on intact cells23. The interaction of CIGB-300 with the holoenzyme may disturb its enzymatic activity, and promote subsequent subunit degradation40. In this vein, we observed in NCI-H522 cells clear reductions of the three CK2 subunits upon CIGB-300 incubation; whereas, slight decreases of CK2α1 in A549 (LUAD) and CK2α2 in NCI-H226 (LUSC) were also noted. As expected, downstream CK2 phosphorylation events were impaired, being CDC37s13 and S6s235/236 the most consistent across the four cell lines tested. In addition, CIGB-300-NPM1 interaction and the subsequent NPM1s125 phosphorylation inhibition were also confirmed. This go along with the partial nucleolar localization and the ribosome biogenesis impairment observed at 3 and 6 h, particularly in LUSC cells. In summary, both direct enzyme interaction/inhibition and phosphoaceptor site binding/ blockage may account in evaluated LUAD and LUSC cells as described before in other cellular models20,22,23.
On time with uptake (0–30 min), target engagement (45 min) and CK2-mediated phosphorylation impairment (1–6 h), CIGB-300 induces mitochondria depolarization (1–6 h) and ROS production (6–24 h) in both NSCLC tumor types. Considering that NAC counteracts to some extent the subsequent cell death, we believe ROS production is partially involved in CIGB-300’s mediated cell killing. Mitochondria depolarization upon CK2 inhibition has been connected before to the intrinsic apoptotic pathway41. In addition, our group and others has reported that CK2 inhibition in NSCLC induce cell death by apoptosis31,42,43,44. Herein we could not confirm an apoptotic cell death on CIGB-300’s incubated cell by using three different markers which otherwise render inconclusive evidences (AnnexinV, PARP, Caspase-3).
In summary, our in vitro results suggest that CIGB-300 by targeting CK2-mediated phosphorylation through a so called dual-mechanism of action, impacts nuclear and cytosolic processes which rely on CK2 activity, thus triggering a still-to-be-characterized cell death pathway in cell lines of LUAD and LUSC origin. This notion is also supported by our former proteomics and genomics studies in the adenosquamous cell line NCI-H12529,45. In addition, tumor cell penetration by CIGB-300 was confirmed here as a key barrier to overcome with a second-generation peptide in order to achieve drug potency in the low micro- or nano-molar range.
To increase the translational power of our research, we used the NCI-H226 cell-line based tumor xenograft to evaluate the anti-tumor effect of CIGB-300 when administered by intravenous route. In line with in vitro experiments, CIGB-300 caused a significant tumor growth delay and increased the survival of tumor bearing-mice, particularly at 10 mg/kg of peptide. Albeit a limited in vivo exploratory study, we observed this peptide dose decreased CK2ɑ1 and RPS6s235/236 levels in the tumor mass. Altogether, our findings further encourage the in vivo evaluation of putative CIGB-300 pharmacodynamic biomarkers hinted before: RPS6s235/236, NPM1s125, CK2ɑ1, CK2ɑ2, CDC37s13 and AKTs12924,29,46; however, also suggest that such biomarkers could be tumor-type specific (e.g. NPM1s125, CK2ɑ1, CK2ɑ2) or more general (e.g. S6s235/236; CDC37s13) within NSCLC specimens. To note, since we used a limited cohort of cell lines here (n = 2, each NSCLC type), we did not attempt to make generalizations across our data concerning LUAD or LUSC tumors. Instead, we aimed to demonstrate the antineoplastic effect of CIGB-300 in pre-clinical models of NSCLC with fewer therapeutic options, while exploring convergences on the mechanism of action and molecular events thereof.
Conclusions
CK2 inhibition is an emerging topic in NSCLC treatment, particularly its combination potential with primary and adjuvant therapies. As an ubiquitous, pleiotropic, and mostly constitutive actively enzyme, CK2 provides cell-intrinsic and extrinsic cues in supporting malignant transformation47. Thus, the mainstream notion is to combine CK2 inhibitors with current radiotherapy, chemotherapy and immunotherapies to enhance their therapeutic effect and/or tackle tumor resistance12,48,49. Given its already proven safety profile in humans, CIGB-300 become an expedite option to validate the harnessing capacity of CK2 inhibitors over therapies aiming at NSCLC with limited therapeutic options.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Wang, M., Herbst, R. S. & Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 27, 1345–1356 (2021).
Dang, C. V., Reddy, E. P., Shokat, K. M. & Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508 (2017).
Lipinski, K. A. et al. Cancer evolution and the limits of predictability in precision cancer medicine. Trends Cancer 2, 49–63 (2016).
Rivera-Concepcion, J., Uprety, D. & Adjei, A. A. Challenges in the use of targeted therapies in non-small cell lung cancer. Cancer Res. Treat. 54, 315–329 (2022).
Pirker, R. Chemotherapy remains a cornerstone in the treatment of nonsmall cell lung cancer. Curr. Opin. Oncol. 32, 63–67 (2020).
Ortega, C. E., Seidner, Y. & Dominguez, I. Mining CK2 in cancer. PLoS One 9, e115609 (2014).
Chua, M. M. J. et al. CK2 in cancer: Cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals (Basel). 10 (2017).
Litchfield, D. W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 369, 1–15 (2003).
Turowec, J. P., Vilk, G., Gabriel, M. & Litchfield, D. W. Characterizing the convergence of protein kinase CK2 and caspase-3 reveals isoform-specific phosphorylation of caspase-3 by CK2α’: Implications for pathological roles of CK2 in promoting cancer cell survival. Oncotarget 4, 560–571 (2013).
Deshiere, A. et al. Unbalanced expression of CK2 kinase subunits is sufficient to drive epithelial-to-mesenchymal transition by Snail1 induction. Oncogene 32, 1373–1383 (2013).
Yaylim, I. & Isbir, T. Enhanced casein kinase II (CK II) activity in human lung tumours. Anticancer Res. 22, 215–218 (2002).
Li, Q. et al. Association of protein kinase CK2 inhibition with cellular radiosensitivity of non-small cell lung cancer. Sci. Rep. 7, 16134 (2017).
Liu, Y. et al. CK2α’ drives lung cancer metastasis by targeting BRMS1 nuclear export and degradation. Cancer Res. 76, 2675–2686 (2016).
Iegre, J. et al. Chemical probes targeting the kinase CK2: A journey outside the catalytic box. Org. Biomol. Chem. 19, 4380–4396 (2021).
Solares, A. M. et al. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 9, 146 (2009).
Siddiqui-Jain, A. et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 70, 10288–10298 (2010).
Perea, S. E. et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 64, 7127–7129 (2004).
Perera, Y. et al. Systemic administration of a peptide that impairs the protein kinase (CK2) phosphorylation reduces solid tumor growth in mice. Int. J. cancer 122, 57–62 (2008).
Gottardo, M. F. et al. Preclinical efficacy of CIGB-300, an anti-CK2 peptide, on breast cancer metastasic colonization. Sci. Rep. 10, 14689 (2020).
Perera, Y. et al. Clinical-grade peptide-based inhibition of CK2 blocks viability and proliferation of T-ALL cells and counteracts IL-7 stimulation and stromal support. Cancers (Basel). 12 (2020).
Martins, L. R. et al. Targeting chronic lymphocytic leukemia using CIGB-300, a clinical-stage CK2-specific cell-permeable peptide inhibitor. Oncotarget 5, 258–263 (2014).
Rosales, M. et al. Targeting of protein kinase CK2 in acute myeloid leukemia cells using the clinical-grade synthetic-peptide CIGB-300. Biomedicines. 9 (2021).
Perera, Y. et al. CIGB-300 anticancer peptide regulates the protein kinase CK2-dependent phosphoproteome. Mol. Cell. Biochem. 470, 63–75 (2020).
Pérez, G. V et al. CIGB-300 anticancer peptide differentially interacts with CK2 subunits and regulates specific signaling mediators in a highly sensitive large cell lung carcinoma cell model. Biomedicines. 11 (2022).
Farina, H. G. et al. CIGB-300, a proapoptotic peptide, inhibits angiogenesis in vitro and in vivo. Exp. Cell Res. 317, 1677–1688 (2011).
Benavent Acero, F. et al. CIGB-300, an anti-CK2 peptide, inhibits angiogenesis, tumor cell invasion and metastasis in lung cancer models. Lung Cancer 107, 14–21 (2017).
Benavent Acero, F. R. et al. Mechanisms of cellular uptake, intracellular transportation, and degradation of CIGB-300, a Tat-conjugated peptide, in tumor cell lines. Mol. Pharm. 11, 1798–1807 (2014).
Gazdar, A. F., Girard, L., Lockwood, W. W., Lam, W. L. & Minna, J. D. Lung cancer cell lines as tools for biomedical discovery and research. J. Natl. Cancer Inst. 102, 1310–1321 (2010).
Perera, Y. et al. Pharmacologic inhibition of the CK2-mediated phosphorylation of B23/NPM in cancer cells selectively modulates genes related to protein synthesis, energetic metabolism, and ribosomal biogenesis. Mol. Cell. Biochem. 404, 103–112 (2015).
Vázquez-Blomquist, D. et al. Gene expression profiling unveils the temporal dynamics of CIGB-300-regulated transcriptome in AML cell lines. BMC Genomics 24, 373 (2023).
Perera, Y. et al. Sensitivity of tumor cells towards CIGB-300 anticancer peptide relies on its nucleolar localization. J. Pept. Sci. Off Publ. Eur. Pept. Soc. 18, 215–223 (2012).
Rosales, M. et al. CIGB-300-regulated proteome reveals common and tailored response patterns of AML cells to CK2 inhibition. Front. Mol. Biosci. 9, 834814 (2022).
Sondka, Z. et al. COSMIC: A curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 52, D1210–D1217 (2024).
Barretina, J. et al. The cancer cell line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. Off Publ. Eur. Pept. Soc. 20, 760–784 (2014).
Skotland, T., Iversen, T. G., Torgersen, M. L. & Sandvig, K. Cell-penetrating peptides: Possibilities and challenges for drug delivery in vitro and in vivo. Molecules 20, 13313–13323 (2015).
Stauber, R. H. & Pavlakis, G. N. Intracellular trafficking and interactions of the HIV-1 Tat protein. Virology 252, 126–136 (1998).
Ruseska, I. & Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 11, 101–123 (2020).
Zhou, Y., Du, W., Koretsky, T., Bagby, G. C. & Pang, Q. TAT-mediated intracellular delivery of NPM-derived peptide induces apoptosis in leukemic cells and suppresses leukemogenesis in mice. Blood 112, 2474–2483 (2008).
Borgo, C. et al. Generation and quantitative proteomics analysis of CK2α/α’((-/-)) cells. Sci. Rep. 7, 42409 (2017).
Qaiser, F. et al. Protein kinase CK2 inhibition induces cell death via early impact on mitochondrial function. J. Cell. Biochem. 115, 2103–2115 (2014).
Yang, B., Yao, J., Li, B., Shao, G. & Cui, Y. Inhibition of protein kinase CK2 sensitizes non-small cell lung cancer cells to cisplatin via upregulation of PML. Mol. Cell. Biochem. 436, 87–97 (2017).
Bliesath, J. et al. Combined inhibition of EGFR and CK2 augments the attenuation of PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer Lett. 322, 113–118 (2012).
So, K. S. et al. AKT/mTOR down-regulation by CX-4945, a CK2 inhibitor, promotes apoptosis in chemorefractory non-small cell lung cancer cells. Anticancer Res. 35, 1537–1542 (2015).
Rodríguez-Ulloa, A. et al. Proteomic profile regulated by the anticancer peptide CIGB-300 in non-small cell lung cancer (NSCLC) cells. J. Proteome Res. 9, 5473–5483 (2010).
Perera, Y. et al. Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity. Mol. Cancer Ther. 8, 1189–1196 (2009).
Firnau, M.-B. & Brieger, A. CK2 and the hallmarks of cancer. Biomedicines. 10 (2022).
D’Amore, C., Borgo, C., Sarno, S. & Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy - potential clinical relevance. Cell. Oncol. (Dordr) 43, 1003–1016 (2020).
Gou, Q. et al. Inhibition of CK2/ING4 pathway facilitates non-small cell lung cancer immunotherapy. Adv. Sci. 10 e2304068 (Weinheim, Baden-Wurttemberg, Ger., 2023).
Acknowledgements
This research was supported by the “National key R&D program of China 2021YFE0192100”.
Author information
Authors and Affiliations
Contributions
Y.Y. Investigation L. D. InvestigationY. L. Methodology, InvestigationC. T. InvestigationDM. V. Methodology; InvestigationG. Z. Methodology; InvestigationD. J. Methodology; SupervisionK. Y. Funding acquisition; Project administration; ResourcesSE. P. Funding acquisition; Investigation; SupervisionY. P. Conceptualization; Methodology; Investigation; Writing - original draft; Funding acquisition; Project administration.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All procedures were operated according to the Guide for the Care and Use of Laboratory Animals (Eighth Edition) issued by The National Academics (Washington, DC), Animal Ethics Committee of Hunan Provincial Research Center for Safety Evaluation of Drugs and ARRIVE Guidelines (https://arriveguidelines.org/).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yi, Y., Dai, L., Lan, Y. et al. CIGB-300 internalizes and impairs viability of NSCLC cells lacking actionable targets by inhibiting casein kinase-2 signaling. Sci Rep 14, 26038 (2024). https://doi.org/10.1038/s41598-024-75990-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-75990-1
