
Abstract
High SARS-CoV-2-specific antibody levels can protect against SARS-CoV-2 reinfection. The gut microbiome can affect a host’s immune response. However, its role in the antibody response to SARS-CoV-2 in people living with HIV (PLWH) remains poorly understood. Here, we categorised PLWH and healthy individuals into high- and low-antibody-response groups. Shotgun metagenomic sequencing and targeted metabolomic assays were used to investigate the differences in the gut microbiome and metabolic functions between the high- and low-antibody-response groups. PLWH demonstrated a higher abundance of short-chain fatty acid (SCFA)-producing species, accompanied by high serum levels of several SCFAs, in the high-antibody-response group than in the low-antibody-response group. In contrast, healthy individuals demonstrated higher enrichment of pilus-bearing bacterial species, with flagella-expressing genes, in the high-antibody-response group than in the low-antibody-response group. Therefore, gut-microbiota-derived SCFAs play a key role in antibody responses in PLWH but not in healthy individuals. Our results afford a novel understanding of how the gut microbiome and its metabolites are associated with host immunity. Moreover, they may facilitate the exploration of modalities to prevent SARS-CoV-2 reinfection through various gut-microbiota-targeted interventions tailored to different populations.
Similar content being viewed by others


Introduction
COVID-19, caused by SARS-CoV-2, emerged in 20191. As of December 31, 2023, the World Health Organization had reported 773,819,856 confirmed COVID-19 cases, including 7,010,568 deaths2. Despite prior COVID-19 exposure diminishing the risk of subsequent infection considerably, cases of SARS-CoV-2 reinfection within 8 postinfection months have been noted3. The presence of SARS-CoV-2-specific antibodies is crucial for COVID-19 pathogenesis and for providing protection against reinfection4,5,6,7.
Trillions of microorganisms, including bacteria, fungi, viruses, and other life forms, reside within the human intestines, forming the gut microbiome. These microorganisms play critical roles in the development of the innate and adaptive immune systems in hosts8. Respiratory viral infections might be associated with alterations in the gut microbiome, which predisposes patients to secondary bacterial infections; this interaction between the gut microbiome and lung health is called the gut-lung axis9. One of the gut-lung axis communication pathways elicits direct effects. For instance, peptidoglycans and lipopolysaccharides (LPSs) from the gut microbiome can enhance the host antibody responses through toll-like receptor (TLR) 4. The gut microbiome can also affect lung health through indirect effects on host immune. Possible mechanisms include short-chain fatty acid (SCFA) production, extraintestinal T-cell population regulation, immune tolerance development through regulatory T cells (Tregs), and systemic inflammation regulation9,10,11,12. The potential significance of intricate gut-lung communication could play a role in determining the antibody response and the vulnerability of airways to SARS-CoV-2 infection, as well as clinical outcomes of COVID-19, which are influenced influenced by individual variations in microbiota composition and diversity13,14. Gut dysbiosis increases an individual’s susceptibility to COVID-19-associated complications13,14. By the end of 2018, the number of people living with HIV (PLWH) globally was estimated to be 37.9 million individuals; they were considered to have impaired immunity and gut dysbiosis, as well as be susceptible to severe COVID-1915–17.
Several studies have reported that despite having lower immune levels, PLWH exhibit SARS-CoV-2-specific antibody response levels comparable to those in healthy individuals following SARS-CoV-2 infection or vaccination18,19,20. However, the mechanisms through which host gut microbiome composition influences immune responses to COVID-19 in PLWH with gut dysbiosis and in healthy individuals with gut homeostasis remain unclear. The comprehension of this phenomenon may enhance the current understanding of the intricate interplay between a host and their gut microbiome; it may also provide insights into strategies for enhancing humoral immunity and protecting against SARS-CoV-2 reinfection in individuals with gut homeostasis and those with gut dysbiosis.
In this study, PLWH and healthy individuals were recruited and followed up until 3 months after SARS-CoV-2 infection. Shotgun metagenomic sequencing and targeted metabolomic assays were performed to examine the roles of the gut microbiome and metabolism in humoral immune responses against SARS-CoV-2 infection. The current study is the first to demonstrate the mechanisms through which various gut microbiome compositions during gut dysbiosis and homeostasis interact with immune responses to SARS-CoV-2 infection differently. The results may provide a comprehensive understanding of the associations of gut microbiome function with immune responses in hosts, thus broadly reducing SARS-CoV-2 reinfection risk.
Results
Study participants and clinical characteristics
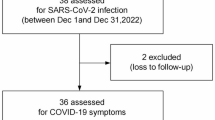
In total, 122 PLWH and 148 healthy individuals with confirmed SARS-CoV-2 infection for the first time were enrolled in the analysis (Fig. 1). Their serum samples were collected 3 months after the infection and analysed using enzyme-linked immunosorbent assay (ELISA). We divided the healthy individuals and PLWH into high-antibody-response (HR) and low-antibody-response (LR) groups according to their ELISA results. The HR group included participants with OD450 for anti-Omicron BA.1-S IgG1 (1:10,000) of > 2 and OD450 for anti-Omicron BA.1-RBD IgG (1:10,000) of > 1, whereas the LR group comprised participants with OD450 for anti-Omicron BA.1-S IgG1 (1:10,000) of < 0.5 and OD450 for anti-Omicron BA.1-RBD IgG (1:10,000) of < 1.

Flow of sample collection and analyses.
In total, 122 serum and 78 faecal samples were collected at baseline from the PLWH group participants confirmed to have SARS-CoV-2 infection. Of the 122 PLWH with baseline serum samples, 37 each were included in the PLWH_HR and PLWH_LR groups, respectively. Table S1 compares the characteristics of participants in the PLWH_LR and PLWH_HR groups based on the baseline blood samples. The PLWH_HR group participants demonstrated higher red blood cell-RBD levels, haemoglobin levels, haematocrit levels, natural killer (NK) cell counts, total B-cell counts and percentages, CD4+/CD8 + ratios, CD4 + T-cell counts, albumin levels, Na levels, and Cl levels. In contrast, the PLWH_LR group had more participants with a viral load of > 50 copies/mL, higher basophil counts, red blood cell distribution width-coefficient of variation (RDW-CV) values, and CD8 + T-cell percentages.
In total, 148 serum and 140 faecal samples were collected at baseline from the HC group participants confirmed to have SARS-CoV-2 infection. Of the 148 healthy individuals with baseline serum samples, 37 and 30 were included in the HC_LR and HC_HR groups, respectively. Table S2 compares the characteristics of HC_LR and HC_HR group participants with baseline serum samples. The HC_LR group participants demonstrated higher basophil counts and percentages, whereas the HC_HR group participants demonstrated higher uric acid levels. In general, these results demonstrate that high red blood cell-RBD levels, haemoglobin levels, haematocrit levels, NK cell counts, total B-cell counts and percentages, CD4+/CD8 + ratios, CD4 + T-cell counts, albumin levels, Na levels, and Cl levels may be positively correlated with high antibody response to SARS-CoV-2 in PLWH and that high basophil counts may be positively correlated with low antibody response to SARS-CoV-2 in PLWH and healthy individuals. In addition, PLWH with a viral load of > 50 copies/mL were more likely to have a lower antibody response to SARS-CoV-2.
Stool samples were collected from 78 patients in the PLWH group and 140 individuals in the HC group at baseline. These samples were subjected to metagenomic sequencing and categorised into PLWH_HR (n = 17), PLWH_LR (n = 25), HC_HR (n = 22), and HC_LR (n = 37) groups (Fig. 1); however, nine of these samples were excluded because of sequencing data shortage and low read number, respectively. Finally, 16, 24, 21, and 31 samples from the PLWH_HR, PLWH_HR, HC_LR, and HC_LR groups were used in the subsequent analyses, respectively (Fig. 1); Tables S3 and S4 list the clinical characteristics of these groups. Of the participants included in metagenomic sequencing, the PLWH_HR group demonstrated considerably higher anti-Omicron BA.1-S IgG, IgG1, IgG2, and IgA levels, as well as anti-Omicron BA.1-RBD IgG, IgG1, and IgG2 levels, than the PLWH_LR group. However, no significant differences were noted between the PLWH_HR and PLWH_LR groups for the following antibodies (Mann–Whitney U test; Fig. 2A): anti-Omicron BA.1-S IgM; anti-Omicron BA.1-RBD IgA, and IgM; and anti-Omicron BA.1-N IgG. In the HC group involved in the metagenomic sequencing, except for anti-Omicron BA.1-RBD IgM, all anti-Omicron BA.1 protein-specific antibodies demonstrated considerably higher levels in the HC_HR group than in the HC_LR group; the between-group differences in anti-Omicron BA.1-RBD IgM levels were nonsignificant (Mann–Whitney U test; Fig. 3A).

Differences in the Omicron BA.1 protein-specific antibody levels and gut microbiome between the PLWH_LR and PLWH_HR groups used for metagenomic sequencing. (A) Omicron BA.1 protein-specific antibody levels as illustrated by OD450 values. Comparisons between PLWH_HR and PLWH_LR groups were made using the Wilcoxon rank-sum test. (B) Difference in beta diversity (in PCoA plots based on Bray-Curtis) between the PLWH_LR and PLWH_HR groups. P values are obtained using PERMANOVA. (C, D) Relative abundance of bacterial phyla (C) and species (D) based on median values. (E) LEfSe analysis for key discriminative species with LDA scores on the log10 scale > 2 between the two groups. PLWH_LR, n = 24; PLWH_HR, n = 16; ns, P > 0.05; ****P < 0.0001, ***P < 0.001; **P < 0.01; *P < 0.05.
We also evaluated the levels of antibodies against wild type (WT), Omicron BA.5, and Omicron XBB.1 proteins between the HC_HR and HC_LR groups as well as between the PLWH_HR and PLWH_LR groups for those included in metagenomic sequencing. The levels of the following antibodies were considerably higher in the PLWH_HR group than in the PLWH_LR group: anti-WT-S, anti-WT-RBD, and anti-WT-N IgGs; anti-Omicron BA.5-S IgG and IgA; anti-Omicron BA.5-RBD IgG and IgA; and anti-Omicron XBB.1-S IgG, IgG1, and IgA; and anti-Omicron XBB.1-RBD IgG, IgG1, and IgA (Mann–Whitney U test; Fig. S1A). Similar results were noted for the HC_LR and HC_HR groups (Fig. S1B).

Differences in the Omicron BA.1 protein-specific antibody levels and gut microbiome between the HC_LR and HC_HRgroups used for metagenomic sequencing. (A) Omicron BA.1 protein-specific antibody levels, as indicated by OD450values. Comparisons between the PLWH_HR and PLWH_LR groups were made using the Wilcoxon rank-sum test. (B) Betadiversity differences (illustrated by PCoA plots based on Bray-Curtis dissimilarity) between the HC_LR and HC_HR groups. Pvalues were determined using PERMANOVA. (C, D) Relative abundance of bacterial phyla (C) and species ( D) based onmedian values. (E) LEfSe analysis for key discriminative species, with LDA scores on the log10 scale > 2, between the twogroups. HC_LR, n = 31; HC_HR, n = 21; ns, P > 0.05; ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
Influence of gut microbiome profile on antibody response to SARS-CoV-2 infection via SCFAs in PLWH
To characterise the effects of gut microbiome composition and function on the antibody response to SARS-CoV-2 infection in PLWH, we analysed the difference in gut microbiome between the PLWH_LR and PLWH_HR groups through shotgun metagenomic sequencing. The PLWH_LR and PLWH_HR groups demonstrated no significant differences in alpha diversity at any level (Wilcoxon rank-sum test; Fig. S2A) and beta diversity in gut microbial composition [Bray–Curtis distances and principal coordinates analysis (PCoA) plots; Fig. 2B], as well as in gut microbial pathway compositions (Fig. S2C; Maaslin2, P < 0.05 indicated significant difference).
Next, we compared the gut microbiome compositions between the PLWH_HR and PLWH_LR groups at the broad taxonomic levels. Notably, at the phylum level, Proteobacteria demonstrated higher relative abundance, whereas Bacteroidetes demonstrated lower relative abundance in the PLWH_LR group than in the PLWH_HR group. At the species level, the top three species identified were Prevotella copri, Escherichia coli, and Ruminococcus gnavus. Compared with the PLWH_LR group, the PLWH_HR group demonstrated significantly higher P. copri abundance but significantly lower E. coli abundance (Fig. 2C, D). Next, we assessed the differentially abundant potential biomarkers contributing to high antibody response to SARS-CoV-2 through LEfSe analysis. The results indicated that the PLWH_HR group was enriched in SCFA-producing species, such as Firmicutes bacteria (including Faecalibacterium, Phascolarctobacterium faecium, and Coprobacillus), but the PLWH_LR group was not (Fig. 2E).
We then investigated the correlation between these species in the PLWH_LR and PLWH_HR groups and the levels of antibodies against WT, Omicron BA.1, Omicron BA.5, and Omicron XBB.1 proteins. Similar to the above results, we observed that the abundance of SCFA-producing species, such as Phascolarctobacterium, Faecalibacterium, Coprobacillus, and Blautia, were positively associated with the levels of anti-Omicron BA.1-S IgG, IgG1, IgG2, and IgA and anti-Omicron BA.1-RBD IgG, IgG1, and IgG2 (Fig. 4A). Moreover, these species were positively associated with the levels of antibodies against WT, Omicron BA.5, and Omicron XBB.1 proteins (Fig. 4A).

Relationship between the gut microbiome, SCFAs, and SARS-CoV-2 antibody response in the PLWH group involved in metagenomic sequencing. (A) Heatmap of Spearman correlation between the key discriminative species relative abundance in the PLWH_LR and PLWH_HR groups, and anti-SARS-CoV-2 protein-specific antibody levels in the PLWH group. Species for this analysis were selected through LEfSe. (B) Heatmap of Spearman correlation between serum SCFA levels and anti-SARS-CoV-2 protein-specific antibody levels in the PLWH group. (C) Heatmap of Spearman correlation between the key discriminative species relative abundance and serum SCFAs levels in the PLWH group. Species for this analysis were selected through LEfSe. (D, E) Boxplots comparing serum isobutyric (D) and propionic (E) acid levels between the high and low antibody responders in the HC and PLWH groups. Comparisons between subgroups were performed using the Mann–Whitney U test. (F) Correlation of serum isobutyric acid levels with anti-Omicron S IgG levels in the PLWH group. P values were determined using the Spearman correlation analysis (n = 40). (G) Correlation of serum propionic acid levels with anti-BA.5 S IgG levels in the PLWH group. P values were determined using the Spearman correlation analysis (n = 40). HC_LR, n = 31; HC_HR, n = 21; PLWH, n = 40; PLWH_LR, n = 24; PLWH_HR, n = 16. ns, P > 0.05; **P < 0.01; *P < 0.05.
Considering the results mentioned above, we used targeted metabolic analysis to verify the difference in serum metabolites between the HR and LR groups (Fig. 4D and E and Fig. S3). The results indicated that the levels of SCFAs, including isobutyric and propionic acids, were considerably higher in the PLWH_HR group than in the PLWH_LR group. Notably, isobutyric and propionic acids were also positively associated with the levels of antibodies against Omicron BA.1-S or Omicron BA.1-RBD (Fig. 4B, F and G). Moreover, propionic acid was positively associated with anti-Omicron BA.5-S, anti-Omicron BA.5-RBD, and anti-Omicron XBB.1-RBD IgG levels (Fig. 4B). Furthermore, SCFA-producing species, such as Faecalibacterium, were found to be positively associated with isobutyric and propionic acid levels. More bacterial species positively associated with SCFAs were enriched in the PLWH_HR group than in the PLWH_LR group (Fig. 4C). These findings suggest that gut microbe-mediated SCFAs may be the drivers of antibody response to COVID-19 in PLWH with gut dysbiosis.
Association of the gut microbiome profile with antibody response to SARS-CoV-2 infection in healthy individuals
As noted above, the gut microbiome may be associated with an increase in SCFA-mediated humoral immune response in PLWH. However, the effects of the gut microbiome on the humoral immunity of healthy individuals remain unclear, mainly because of considerable differences in the gut microbiome between PLWH and healthy individuals, as well as the long-term immune dysfunction of their gut microbiome experienced by PLWH21.
In this study, the gut microbiomes were found to demonstrate significant differences between the PLWH and HC groups. Alpha diversity was considerably higher in the HC group than in the PLWH group (Fig. S4A-D). A permutational multivariate analysis of variance (PERMANOVA) of beta diversity, based on Bray-Curtis distances, revealed that bacterial communities differed significantly between the PLWH and HC groups (P = 0.001; Fig. S4E). In total, 54 bacterial taxa were noted to differ between the PLWH and HC groups, with log10-transformed linear discriminant analysis (LDA) score = 3.0 (Fig. S4F). Many functions of the PLWH group, including flagellar assembly, lysine degradation, beta-lactam resistance, DNA replication, and lipoic acid metabolism, were also found to be depleted in the PLWH group compared with the HC group (Fig. S5). However, the levels of SARS-CoV-2-specific antibodies did not differ significantly between the two groups (Fig. S6)—consistent with previous reports20.
Considering gut microbiome dysbiosis in PLWH, we also analysed the association between gut microbiome and immune response in healthy individuals. The differences in alpha diversity between the HC_LR and HC_HR groups were nonsignificant (Fig. S2B). In contrast, PCoA of beta diversity based on Bray-Curtis distances suggested considerable differences between the HC_LR and HC_HR groups (P = 0.024; Fig. 3B). Next, we compared gut microbiome composition between the HC_LR and HC_HR groups and observed that both groups demonstrated an overall dominance of Proteobacteria, Bacteroidetes, and Firmicutes at the phylum level—similar to that noted in the PLWH subgroups. However, the HC_HR group demonstrated lower Firmicutes abundance but higher Proteobacteria and Bacteroidetes abundance than the HC_LR group (Fig. 3C). At the species level, the HC_HR group demonstrated a higher E. coli and Phocaeicola vulgatus abundance but less Ruminococcaceae bacterium, P. copri, and Faecalibacterium prausnitzii abundance than the HC_LR group (Fig. 3D). Next, we used LEfSe to identify the taxa with significantly different abundance between the HC_LR and HC_HR groups further. The results indicated that the relative abundance of E. coli, K. pneumoniae, S. enterica, Clostridium nexile CAG:348, Paraprevotella clara CAG:116, and Ruminococcus sp. CAG:177 was significantly higher in the HC_HR group than in the HC_LR group (Fig. 3E).
We then investigated the correlation of these enriched species with the levels of antibodies against WT, Omicron BA.1, Omicron BA.5, and Omicron XBB.1 proteins in the HC_HR group. Notably, the results indicated that Pa. clara CAG:116, S. enterica, Ruminococcus sp. CAG:177, C. nexile CAG:348, K. pneumonia, and E. coli abundance was positively associated with anti-Omicron BA.1-S and anti-Omicron BA.1-RBD IgG levels. Moreover, the relative abundance of Pa. clara CAG:116 and Ruminococcus sp. CAG:177 was positively associated with the levels of anti-WT-S, anti-WT-RBD, anti-WT-N, anti-Omicron BA.5-S, anti-Omicron BA.5-RBD, anti-Omicron XBB.1-S, and anti-Omicron XBB.1-RBD IgGs, as well as anti-Omicron XBB.1-S IgG1. Moreover, the relative abundance of S. enterica, C. nexile CAG:348, K. pneumoniae, and E. coli was positively associated with the levels of anti-Omicron BA.1-RBD IgM, IgA, and IgG2, as well as anti-Omicron BA.1-S IgM (Fig. S7).
We further identified bacterial components likely expressed by the species enriched differently between the HC_LR and HC_HR groups. According to Gene Ontology (GO) annotation, Ruminococcus sp. CAG:177 and Pa. clara CAG:116 were positively associated with the cell-cell junction (GO: 0005911) and zymogen granule membrane (GO: 0042589), respectively. Moreover, E. coli, S. enterica, K. pneumoniae, and C. nexile CAG:348 were positively associated with cell components, such as the bacterial pilus (GO: 0009289), cell division site (GO: 0032153), cell septum (GO: 0030428), and host cell wall (GO: 0044158) (Fig. S8). Interestingly, E. coli, S. enterica, K. pneumoniae, and C. nexile CAG:348 enriched in high antibody responders (Fig. 3E) were more likely to demonstrate flagella gene expression (Fig. 5C-F). The relative abundance of E. coli, S. enterica, and K. pneumonia was higher than that of the other species enriched in high antibody responders (Fig. 5A). Moreover, E. coli contributed mostly to the bacterial pilus (Fig. 5A and C; Fig. S9). Moreover, bacterial pilus (GO: 0009289), cell division site (GO: 0032153), cell septum (GO: 0030428), host cell wall (GO: 0044158), and zymogen granule membrane (GO:0042589) levels were higher in the HC_HR group than in the HC_LR group. Of all these cell components, the bacterial pilus (GO: 0009289) demonstrated the highest relative abundance (Fig. 5B; Fig. S9).

Relationship of relative abundance of the bacterial pilus with that relative abundance of E. coli, K. pneumonia, S. enterica, and C. nexile CAG:348, and its difference between the HC_LR and HC_HR groups. (A) Heatmap of relative abundance of the key discriminative species between the HC_LR and HC_HR groups and their contribution to bacterial pilus (GO: 0009289). Red text denotes species enriched in the HC_HR group. Correlation between pilus gene abundance and species relative abundance was examined using the Spearman correlation analysis. (B) Comparison of the relative abundance of the bacterial pilus (GO: 0009289) in the HC_HR and HC_LR groups. Comparison between the HC_LR and HC_HR groups was made using the Wilcoxon rank-sum test. (C - F) Comparison of the relative abundance of E. coli (C), K. pneumonia (D), S. enterica (E), and C. nexile CAG:348 (F) in the HC_HR and HC_LR groups. The bacterial abundance was positively associated with the abundance of the bacterial pilus (GO: 0009289). Comparisons between HR and LR groups and between the highest tier and other tiers were made using the Wilcoxon rank-sum test. HC, n = 52; HC_LR, n = 31; HC_HR, n = 21; ***P < 0.001; **P < 0.01; *P < 0.05.
Notable differences were observed in KEGG level 3 pathway compositions between the HC_HR and HC_LR groups (Fig. S10A). In the HC_HR group, seven microbial pathways (e.g. pathogenic E. coli infection pathway) demonstrated more enrichment, whereas eight microbial pathways demonstrated less enrichment. Moreover, the pathogenic E. coli infection pathway was positively associated with anti-Omicron BA.1-S IgG, anti-Omicron BA.1-RBD IgA, anti-Omicron BA.1-N IgG, anti-WT-S IgG, and anti-Omicron XBB.1-RBD IgA levels (Fig. S10B). Furthermore, E. coli, S. enterica, K. pneumoniae, and C. nexile CAG:348—but not Ruminococcus sp. CAG:177 and Pa. clara CAG:116—were noted to have the highest enrichment of function pathways in the HC_HR group (Fig. S10C).
SCFAs, including isobutyric and propionic acids, were associated with the high antibody response to SARS-CoV-2 infection in PLWH. Thus, we next assessed whether SCFAs have similar effects in healthy individuals. The results indicated that acetic acid levels were higher in the HC_LR group than in the HC_HR group; however, we noted no significant differences in the levels of other SCFAs between the two groups (Fig. 4D and E; Fig. S3).
Finally, we used binary logistic regression and generated predictive models to assess whether the gut microbes enriched in the HC_HR group could be used to classify the high antibody responders from the low antibody responders. Notably, Pa. clara CAG:116, K. pneumoniae, E. coli, Ruminococcus sp. CAG:177, C. nexile CAG:348, and S. enterica and their total relative abundance were selected to establish the predictive models for high antibody response. The results of the Receiver operating characteristic (ROC) curve analysis of these models demonstrated that a combination of the aforementioned six bacterial species had the highest predictive power, with area under the ROC curve (95% confidence interval) = 0.705 (0.555–0.855) (P = 0.013; Fig. S11A and B).
Correlation of clinical characteristics with immune responses in PLWH and healthy individuals
We further investigate the association between the clinical characteristics of and immune response in PLWH (Fig. S12) and healthy individuals (Fig. S13). We found that the CD8 + T-cell percentages, total T-cell percentages, basophil counts, RDW-CV values, mean corpuscular haemoglobin concentrations, monocyte percentages, glucose levels, calcium levels, uric acid levels, aspartate aminotransferase (AST)/alanine aminotransferase (ALT) ratios, albumin/globulin ratios, basophil percentages, and creatinine levels were negatively associated with antibody response in PLWH. Moreover, total B-cell counts, total B-cell percentages, high-density lipoprotein cholesterol (HDL-C), haematocrit levels, hemoglobulin levels, globulin levels, total protein levels, indirect bilirubin levels, CD4 + T-cell counts, NK cell counts, chloride levels, and sodium levels were positively correlated with the antibody response in the PLWH. Ph. faecium and Phascolarctobacterium spp., the bacterial species enriched in the PLWH_HR group, were positively associated with the NK cell counts and percentages (Fig. S14).
In healthy individuals, clinical findings such as mean triglyceride, as well as ALT, corpuscular haemoglobin concentrations, gamma-glutamyl transferase, alkaline phosphatase, and uric acid levels, were positively associated with SARS-CoV-2-specific antibody levels, whereas direct bilirubin levels, basophil percentages, and albumin/globulin ratios were negatively associated with SARS-CoV-2-specific antibody levels (Fig. S13). Notably, HDL-C levels and basophil counts and percentages were positively associated with the species enriched in the LR groups, whereas eosinophil counts and percentages were positively associated with the species enriched in the HR groups (Fig. S15).
Discussion
Since 2019, when the first case of COVID-19 was reported, the disease has become a relatively common infectious disease22; currently, about 15% of the population has experienced SARS-CoV-2 reinfection22,23. Numerous recent studies have revealed that both innate and adaptive immune responses become activated in response to SARS-CoV-2 infection, reducing the numbers of circulating B cells, NK cells, CD4 + cells, CD8 + cells, basophils, eosinophils, and monocytes in COVID-19 patients24. High levels of SARS-CoV-2 protein-specific antibodies protect against multiple infections25. Patients with COVID-19 exhibit an imbalance in their gut microbiome, and its composition is correlated with serum cytokine, chemokine, and inflammation marker levels. The gut microbiome may, therefore, influence the disease severity and host immune response26.
The association between the gut microbiome and host immune system have been investigated extensively27,28. Pathogen-associated molecular patterns, such as flagellin, peptidoglycans, and LPSs, produced by the gut microbiome can activate pattern recognition receptors; therefore, they may contribute to various aspects of host development, including immune response enhancement28,29. Four classes of PRRs have been discovered thus far, including NOD-like receptors, C-type lectin receptors, cytoplasmic proteins (e.g. retinoic acid-inducible gene-I-like receptors), and transmembrane proteins (e.g. TLRs)29. In addition to molecules sensed by PRRs, the gut microbiome produces many metabolites with the potential to modulate immune responses30. Among these metabolites, SCFAs, such as acetic, butyric, and propionic acids, have been studied the most10,29. SCFAs are the main metabolic end products of bacterial fermentation in the colon. They enhance B-cell metabolism by promoting oxidative phosphorylation, glycolysis, and fatty acid synthesis. This metabolic support facilitates optimal homeostatic antibody responses not specific to pathogens and antibody responses during infections10. However, mechanisms through which the gut microbiome modulates immune responses to SARS-CoV-2 in different populations remain poorly understood. In the present study, Chinese HIV-positive and -negative individuals with a recent history of SARS-CoV-2 infection around the end of 2022 were recruited. All participants had received at least two doses of a SARS-CoV-2 vaccine. They were mainly infected with Omicron BA.5.2 and BF.7 variants, as classified by the China Center for Disease Control and Prevention31. The participants were divided into HR and LR groups according to the presence of antibodies against Omicron BA.1-S and Omicron BA.1-RBD approximately 3 months after infection. Moreover, the levels of antibodies against WT, Omicron BA.5, and Omicron XBB.1 proteins were higher in the HR groups than in the LR groups. We, therefore, investigated the between-group differences in the gut microbiome and metabolites through shotgun metagenomic sequencing and targeted metabolomic analyses.
To our knowledge, this is the first human study focused on the effects of gut microbiome composition on immunogenicity for SARS-CoV-2, as well as the potential contributions of the gut microbiome and metabolites to humoral immunity against SARS-CoV-2 in PLWH and healthy individuals. In healthy individuals, higher relative abundance of K. pneumoniae, E. coli, C. nexile CAG:348, S. enterica, Pa. clara CAG:116, and Ruminococcus sp. CAG:177 was associated with higher levels of antibodies against SARS-CoV-2 proteins. Therefore, E. coli, K. pneumoniae, S. enterica, and C. nexile CAG:348 may be an adjuvant for boosting humoral immunity response in healthy individuals. Notably, flagellated bacterial species such as E. coli, S. enterica, and K. pneumonia activate the TLR2 and TLR4 pathways10,32. Moreover, flagellin from these species binds with TLR5, whereas the adhesin portion of their fimbriae can induce the innate immune system via TLR4. Therefore, TLR activation enhances the host immune response to pathogen infection33. Our GO annotation results also indicated that the bacterial pilus, present in E. coli, S. enterica, and K. pneumonia, demonstrated significantly higher expression in the HC_HR group than in the HC_LR group. A similar immunomodulatory function of bacteria with flagella and fimbriae has been noted in individuals who have undergone COVID-19 mRNA34 and seasonal influenza33,35 vaccination. Moreover, the importance of the gut microbiome in stimulating antigen-specific IgG production has been revealed through intranasal immunisation using human serum albumin as a model antigen and cholera toxin as an adjuvant36. In addition, K. pneumoniae, E. coli, and S. enterica were found to be positively associated with IgM, IgA, and IgG2, indicating that a high abundance of these opportunistic pathogens elicits a strong immune response, as well as facilitates chronic infection prevention and inflammation. Furthermore, Pa. clara CAG:116 and Ruminococcus sp. CAG:177 were positively correlated with high levels of all types of antibodies against SARS-CoV-2 proteins, except IgM, IgA, and IgG2.
In PLWH with gut dysbiosis over the long term, the gut microbiome was noted to contribute to the humoral immunity response through SCFAs. Moreover, the gut microbial diversity, composition, and function did not differ significantly between PLWH with high and low antibody responses. However, in PLWH, SCFA-producing microbial species, such as Phascolarctobacterium, Faecalibacterium, Coprobacillus, and Blautia, were significantly more enriched in the HR group than in the LR group. Moreover, SCFAs, including isobutyric and propionic acids, were higher in the HR group than in the LR group. Furthermore, isobutyric and propionic acids were positively correlated with SARS-CoV-2-specific antibody levels. An animal study indicated that microbial-derived SCFAs enhance cellular metabolism and promote -cell differentiation into antibody-producing cells through regulation of gene expression10. However, the effects of SCFAs on antibody responses to pathogen infection warrant further investigation37. The current results indicated that microbe-derived SCFAs are associated with the antibody response to SARS-CoV-2 infection in PLWH with gut dysbiosis but not in healthy individuals with gut homeostasis. Our findings are solely based on an observational study; therefore, further investigation on whether microbe-derived flagella, fimbriae, or SCFAs can differently contribute to antibody response to SARS-CoV-2 during gut dysbiosis and homeostasis is warranted. We also noted that Ph. faecium and Phascolarctobacterium spp.—the species enriched in high antibody responders—were found to be positively associated with NK cell counts and percentages in PLWH. This result confirmed the existence of bidirectional regulation between host immune function and the gut microbiome. HDL-C levels were noted to be positively correlated with the abundance of species associated with low antibody response among healthy individuals. Thus, in hosts, cholesterol metabolism may be linked to gut microbiome composition and subsequently influence immune function. However, this phenomenon requires further investigation.
The present study has several limitations. First, our sample was relatively small. Therefore, the current findings warranted verification in larger cohorts of diverse ethnicities. Second, we did not assess the dynamic changes in the gut microbiome during SARS-CoV-2 infection throughout the course of the disease. Therefore, the current results can only demonstrate the effects of the gut microbiome on immune response in preinfection scenarios. Third, we adjusted for various potential confounders affecting the gut microbial profile; however, we lacked information on diet, medication, and other health conditions, which may affect the gut microbiome and SCFAs. Fourth, all participants received a SARS-CoV-2 vaccine, and the impact of the vaccine on the gut microbiome, SCFAs, and antibody levels could not be controlled. Fifth, there was a sex imbalance between the HC and PLWH groups, and information on sexual practices was not available in this study. This may limit the direct comparability between the groups and could introduce sex-related biases in the results. Finally, we did not use animal models to validate the function of an altered gut microbiome in relation to SARS-CoV-2 immunogenicity.
In summary, we integrated metagenomics and metabolomics to investigate the modulatory function of the gut microbiome and metabolome in humoral immunity to SARS-CoV-2 infection, respectively. Healthy individuals with high antibody response to SARS-CoV-2 were noted to have higher abundance of Pa. clara CAG:116, K. pneumoniae, E. coli, Ruminococcus sp. CAG:177, C. nexile CAG:348, and S. enterica. These enriched species may be biomarkers for distinguishing between patients with high and low antibody levels, thereby predicting reinfection risk. Pilus-bearing bacterial species such as K. pneumoniae, E. coli, C. nexile CAG:348, and S. enterica, which can act as adjuvants by activating immunomodulatory TLR agonists, may play a beneficial role in SARS-CoV-2 immunogenicity. In PLWH, increased SCFA production by the gut microbiome, which could enhance B-cell metabolism, is associated with improved antibody responses to SARS-CoV-2 infection. Moreover, higher levels of microbiome-related SCFAs or specific gut microbes indicate higher levels of antibodies, which may protect against not only the current SARS-CoV-2 strains but also the variants emerging in the future. These novel findings may aid in developing microbiota-targeted interventions to optimise immune responses against SARS-CoV-2 reinfection and enhance the related immunogenicity, thereby protecting populations from SARS-CoV-2 reinfection in varied populations. In the future, clinical research on whether microbiota-targeted interventions in PLWH and healthy individuals optimise immune response to SARS-CoV-2 infection and enhance protection durability is warranted.
Conclusion
In this study, we elucidated mechanisms through which the gut microbiome modulates humoral immunity against SARS-CoV-2 infection differently in PLWH with gut dysbiosis and healthy individuals with gut homeostasis. In healthy individuals with high antibody responses, we noted an enrichment of pilus-bearing bacterial species. In PLWH, the high levels of SCFAs produced by SCFA-producing species were noted to be associated with enhanced antibody responses to SARS-CoV-2 infection. Thus, apart from molecules detected by TLRs, SCFAs derived from the gut microbiota may influence antibody responses, specifically in PLWH rather than in healthy individuals. Finally, the current findings clarify the interactions of the gut microbiome with the immune system and pathogens; they may also provide insights into the pathogeneses of several diseases and facilitate the development of strategies for COVID-19 reinfection prevention.
Methods
Study participants
In total, 130 PLWH (PLWH group) and 158 healthy individuals (HC group) were recruited in this study at the First Affiliated Hospital of Zhejiang University and Zhejiang Qingchun Hospital between February 2022 and November 2022. The exclusion criteria for all participants are as follows: (i) age of < 18 or > 70 years (because the gut microbiome of individuals from these age groups typically differs from that of the general adult population38); (ii) presence of a chronic disease; (iii) antibiotic, probiotic, prebiotic, or symbiotic use within the preceding 6-month period; and (iv) past history of SARS-CoV-2 infection or other conditions making the individuals unsuitable for participation. Their stool, whole blood, and serum samples and their information (e.g. age, sex, and clinical indexes) were collected before SARS-CoV-2 infection (i.e. at baseline). Among these participants, 122 PLWH and 148 healthy individuals developed SARS-CoV-2 infection for the first time between December 2022 and January 2023. SARS-CoV-2 infection was confirmed through IgM-IgG antibody and nucleic acid testing. Serum samples were collected 3 months after their initial SARS-CoV-2 infection. All participants enrolled in this study had received at least two SARS-CoV-2 vaccine doses and demonstrated a mild or moderate disease. Each participant provided written informed consent before participating in the study.
Patients who had had severe COVID-19 were excluded. Here, severe disease was defined according to the Diagnosis and Treatment Plan of COVID-19 (Seventh Edition) and the Novel Coronavirus Pneumonia Prevention and Control Plan (Fourth Edition) (in Chinese, available from: http://www.gov.cn/zhengce/zhengceku/2020-02/07/content_5475813.htm) as follows: (1) shortness of breath (respiratory rate > 30 times/min), (2) resting-state oxygen saturation < 93%, and (3) arterial partial pressure of oxygen/fraction of inspired oxygen < 300 mmHg.
Finally, 122 PLWH and 148 healthy individuals with confirmed SARS-CoV-2 infection were enrolled in this study. Blood samples were collected from 122 PLWH and 144 healthy individuals, whereas faecal samples were collected from 78 PLWH and 140 healthy individuals before SARS-CoV-2 infection onset.
Sample collection and preparation
In total, 140 and 78 fresh stool samples from healthy individuals and PLWH were collected. They were flash-frozen in liquid nitrogen and then stored at -80 °C until analysis. Moreover, 148 and 122 serum samples retrieved for routine biochemical or immunological testing from healthy individuals and PLWH before SARS-CoV-2 infection were collected, respectively. Next, to avoid repeated freezing and thawing, the samples were inactivated at 56 °C for 30 min and dispensed in 100-µL aliquots until later serological analyses. Finally, all aliquoted serum samples were stored at − 80 °C and tested within 3 months of receipt. In total, 122 whole blood samples (5–10 mL) retrieved from PLWH for routine blood testing were used to enumerate lymphocyte subpopulations through flow cytometry immediately after collection. Finally, 148 and 122 serum samples from healthy individuals and PLWH were collected 3 months after SARS-CoV-2 infection to test the levels of SARS-CoV-2-specific antibodies.
Lymphocyte subset analysis on PLWH through flow cytometry
Flow cytometry analysis was performed to quantify various lymphocyte subsets by using Multitest 6-colour TBNK reagent (BD Biosciences), according to the manufacturer’s instructions. In brief, 100 µL of whole blood was incubated with this reagent, followed by lysis using the BD FACS lysing solution. Then, the samples were analysed on a Navios flow cytometer (Beckman Coulter). Lymphocyte subpopulations, including B cells (CD3 − CD19+), T cells (CD3 + CD19−), and NK cells (CD16/CD56+), were differentiated. Within the T-cell subpopulation, additional separation was performed to distinguish CD8 + T cells (CD4 − CD8+) from CD4 + T cells (CD4 + CD8−).
Enzyme-linked immunosorbent assay and patient grouping
ELISA was performed as described previously39. SARS-Cov-2 proteins were purchased from Sino Biological (Beijing, China): full-length spike (S) protein (cat. no. 40589-V08H12), RBD (cat. no. 40592-V08H), and nucleocapsid (N) protein (cat. no. 40588-V07E1) of the WT; S protein (cat. no. 40589-V08H26), RBD (cat. no. 40592-V08H121) and N protein (cat. no. 40588-V07E34) of the BA.1 (Omicron) variant; S protein (cat. no. 40589-V08H32) and RBD (cat. no. 40592-V08H121) of the BA.5 (Omicron) variant; and S protein (cat. no. 40589-V08H42) and RBD (cat. no. 40592-V08H85) of the XBB.1 (Omicron) variant. Next, 96-well plates (Corning, USA) were coated overnight with the aforementioned SARS-Cov-2 proteins at concentrations of 500 ng/mL. After 24 h, these precoated plates were washed three times with phosphate-buffered saline plus 0.1% Tween-20 (PBS-T) and blocked with 1% BSA in PBS-T at room temperature for 2 h. Serum samples were diluted using 1% BSA in PBS-T at a ratio of 1:100 for IgA, IgM, and IgG2 detection and at a ratio of 1:1000 for IgG and IgG1 detection. The diluted serum samples (100 µL) were added to each well, and each sample was assessed in triplicate. Then, the plates were incubated at 37 °C for 90 min, washed three times, and incubated at 37 °C for 1 h with one of the following horseradish peroxidase-conjugated antihuman secondary antibodies (all from Abcam) diluted in 1% BSA in PBS-T: anti-IgG (1:10,000; ab6759), anti-IgG1 (1:2,000; ab99774), anti-IgG2 (1:2,000; ab99779), anti-IgA (1:10,000; ab98558), and anti-IgM (1:10,000; ab97205). Next, the plates were washed three times and then incubated with the 3,3′,5,5′-tetramethylbenzidine substrate at room temperature for 3 min. The reaction was immediately terminated using 1 M H2SO4. The optical density at 450 nm (OD450) of the wells was measured on a microplate reader. To control for plate-to-plate variability, identical positive and negative control samples were included in each plate. For each participant, antibody levels are calculated as mean OD450 values from three identical samples.
DNA extraction, library construction, and metagenomics analysis
Metagenome sequencing and data analysis were performed by OE Biotech (Shanghai, China). Total stool genomic DNA was extracted using a QIAamp fast DNA Stool Mini Kit (Qiagen, Germany), according to the manufacturer’s protocol. Next, DNA concentration was detected through agarose gel electrophoresis and on NanoDrop2000. After purification, DNA was prepared to construct metagenomics shotgun libraries (insert size = 350 bp) by using a TruSeq DNA sample preparation kit (Illumina, San Diego, CA, USA). The prepared DNA library was quantified through quantitative polymerase chain reaction and sequenced on the Illumina NovaSeq 6000 sequencing system in the 150-bp paired-end mode. Six metagenomic HC group samples and two metagenomic PLWH group samples were excluded because of library preparation failure or sequence shortage of < 10 million reads. In total, 92 metagenome samples were included in the analysis (Fig. 1). The raw data were in the FASTQ format. The reads were trimmed and filtered using fastp (version 0.20.1)40. Host pollution control was performed, and the postfiltered pair-end reads were aligned against the host genome by using bbmap (version 38.93-0), and the aligned reads were discarded.
After obtaining valid reads, we performed quality control of the metagenomics data by using MEGAHIT (version 1.2.9)41. We also used Prodigal (version 2.6.3)42 to perform ORF prediction of assembled contigs (> 500 nt) and then translated them into amino acid sequences. The nonredundant sets of genes were constructed for all predicted genes by using MMSeqs2 (version 13.45111)43, with clustering parameters set at 95% identity and 90% coverage. The representative sequence of each gene set was selected as the longest gene. Clean reads from each sample were aligned against the nonredundant gene set (with a threshold of 95% identity) by using salmon (version 1.8.0), and the corresponding sample’s gene abundance information was counted and normalised. DIAMOND (version 0.9.10.1116)44 was used to annotate amino acid sequences of the gene set representatives using the data from the NCBI Nonredundant (nr), Kyoto Encyclopedia of Genes and Genomes, Clusters of Orthologous Genes, SWISS-PROT, and GO databases at an e-value cutoff of 1e − 5. Species taxonomy was determined on the basis of the respective taxonomy database in the NR Library, and species abundance was calculated using gene abundances accordingly and normalised subsequently. To construct abundance profiles at different taxonomic levels, we performed statistical analysis of organism abundance data in terms of ___domain, kingdom, phylum, class, order, family, genus, and species. Table S5 lists all software programs and parameters used here for metagenomic data analysis.
Serum SCFA quantification
Liquid chromatography–mass spectrometry (LC-MS) was used to quantify serum SCFAs, including acetic, propionic, butyric, pentanoic, isobutyric, and isovaleric acids. In brief, the serum samples were thawed; then, 50 µL of a sample was mixed with 50 µL of 400 µL of diethyl ether, 100 µL of the internal standard, and 15% phosphoric acid in a centrifuge tube. Next, this mixture was centrifuged at 135,000 g at 4 °C for 10 min. Then, the supernatant was transferred to a vial for LC-MS analysis.
The LC-MS analysis was performed using an Agilent gas chromatograph connected to an Agilent MS detector equipped with an HP-5MS column. Moreover, high-purity helium was used as the carrier gas at a flow rate of 1 mL/min. Finally, Agilent MSD ChemStation was used to analyse the collected data. To quantify, standard calibration curves were constructed using authentic standards diluted in diethyl ether across concentrations ranging from 0.02 to 500 µg/mL. Method validation included instrument stability, within-day repeatability, accuracy assessment, and interday repeatability.
Statistical analysis
Clinical characteristics and SCFA levels were compared between groups by using the Fisher exact test for the categorical variables and the Wilcoxon rank-sum test for continuous variables. The alpha diversity indexes including Chao1, ACE, and Simpson and Shannon index were compared using the Wilcoxon rank-sum test. We performed Bray–Curtis PCoA by using the Bray-Curtis distance matrix on the QIIME software program. Differential abundance of KEGG level 3 pathways was screened using Microbiome Multivariable Associations with Linear Models (version 2). Differential abundance of species between groups was identified using the LDA effect size (LEfSe version 1.1.01; LDA score > 2; P < 0.05). The bacterial species identified by LEfSe were confirmed using the t test and Wilcoxon rank-sum test. SCFA levels were compared between groups by using the Wilcoxon rank-sum test. Correlation between continuous variables, including anti-SARS-CoV-2-specific antibody levels, species abundance, KEGG level 3 pathway, GO function abundance, SCFA levels, and clinical characteristics, were analysed using the Spearman correlation test. The Wilcoxon rank-sum test was used to assess the between-group differences in continuous variables. Multiple testing was corrected using the Benjamini-Hochberg method to control the false-discovery rate (FDR). P value or FDR-corrected P value < 0.05 was considered to indicate statistical significance. ROC curves were employed to determine the prediction value of the identified biomarkers. All statistical analyses were performed using Statistical Package for the Social Sciences (version 23.0) and GraphPad Prism (version 7.0). Correlation heatmaps with relevant symbols were generated using OmicStudio tools at https://www.omicstudio.cn.
Ethics approval
Statement.
All our human experiments were conducted according to the ethical policies and procedures approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University, China (approval no. IIT20230314B). All the included participants have signed the consent forms.
Data availability
Raw sequence data used for metagenomic sequencing in this study has been uploaded to the National Genomics Data Center (https://ngdc.cncb.ac.cn/gsa/) with primary access codes PRJCA021055. Other data analyzed in this study can be obtained from the corresponding author upon reasonable request.
References
Dong, E., Du, H. & Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 20, 533–534. https://doi.org/10.1016/s1473-3099(20)30120-1 (2020).
WHO. Coronavirus Disease (COVID-19) Situation Dashboard, (2021). https://covid19.who.int/.
Breathnach, A. S. et al. Prior COVID-19 significantly reduces the risk of subsequent infection, but reinfections are seen after eight months. J. Infect. 82, e11–e12. https://doi.org/10.1016/j.jinf.2021.01.005 (2021).
Sanaie, S. et al. Immune response variables and viral mutations impact on COVID-19 reinfection and relapse. Int. Immunopharmacol. 100, 108108. https://doi.org/10.1016/j.intimp.2021.108108 (2021).
Lumley, S. F. et al. Antibody status and incidence of SARS-CoV-2 infection in Health Care workers. N. Engl. J. Med. 384, 533–540. https://doi.org/10.1056/NEJMoa2034545 (2021).
Schäfer, A. et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 218 https://doi.org/10.1084/jem.20201993 (2021).
Scourfield, D. O. et al. The role and uses of antibodies in COVID-19 infections: a living review. Oxf. Open. Immunol. 2, iqab003. https://doi.org/10.1093/oxfimm/iqab003 (2021).
Cresci, G. A. (ed Bawden, E.) Gut microbiome: what we do and don’t know. Nutr. Clin. Practice: Official Publication Am. Soc. Parenter. Enter. Nutr. 30 734–746 https://doi.org/10.1177/0884533615609899 (2015).
Chakradhar, S. A curious connection: teasing apart the link between gut microbes and lung disease. Nat. Med. 23, 402–404. https://doi.org/10.1038/nm0417-402 (2017).
Kim, M., Qie, Y., Park, J. & Kim, C. H. Gut microbial metabolites fuel host antibody responses. Cell. host Microbe. 20, 202–214. https://doi.org/10.1016/j.chom.2016.07.001 (2016).
Ma, P. J., Wang, M. M. & Wang, Y. Gut microbiota: a new insight into lung diseases. Biomed. Pharmacotherapy = Biomedecine Pharmacotherapie. 155, 113810. https://doi.org/10.1016/j.biopha.2022.113810 (2022).
Cait, A. et al. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 11, 785–795. https://doi.org/10.1038/mi.2017.75 (2018).
Wang, M., Zhang, Y., Li, C., Chang, W. & Zhang, L. The relationship between gut microbiota and COVID-19 progression: new insights into immunopathogenesis and treatment. Front. Immunol. 14, 1180336. https://doi.org/10.3389/fimmu.2023.1180336 (2023).
Zhang, F. et al. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat. Reviews Gastroenterol. Hepatol. 20, 323–337. https://doi.org/10.1038/s41575-022-00698-4 (2023).
Kanwugu, O. N. & Adadi, P. HIV/SARS-CoV-2 coinfection: a global perspective. J. Med. Virol. 93, 726–732. https://doi.org/10.1002/jmv.26321 (2021).
Brown, L. B., Spinelli, M. A. & Gandhi, M. The interplay between HIV and COVID-19: summary of the data and responses to date. Curr. Opin. HIV AIDS. 16, 63–73. https://doi.org/10.1097/coh.0000000000000659 (2021).
Ouyang, J. et al. Metformin effect on gut microbiota: insights for HIV-related inflammation. AIDS Res. Therapy. 17 https://doi.org/10.1186/s12981-020-00267-2 (2020).
Fan, X. et al. Safety and Immunogenicity of SARS-CoV-2 vaccines in people living with HIV: a systematic review and Meta-analysis of real-World studies. Infect. Microbes Dis. 5, 159–166 (2023).
Farhadian, N., Sharifi, S., Taghadosi, M., Farhadian, M. & Vaziri, S. Humoral immune response to two doses of COVID-19 mRNA-based vaccines in people living with HIV: a systematic review and meta-analysis. Rev. Med. Virol. 33, e2451. https://doi.org/10.1002/rmv.2451 (2023).
Zhang, X., Han, D., Wu, N. & Serum SARS-CoV-2 antibodies in HIV-1-infected patients after inactivated vaccination and SARS-CoV-2 infection. Heliyon 10, e31731. https://doi.org/10.1016/j.heliyon.2024.e31731 (2024).
Li, S. X. et al. Complexities of gut Microbiome Dysbiosis in the context of HIV infection and antiretroviral therapy. Clin. Pharmacol. Ther. 99, 600–611. https://doi.org/10.1002/cpt.363 (2016).
Willyard, C. Are repeat COVID infections dangerous? What the science says. Nature 616, 650–652. https://doi.org/10.1038/d41586-023-01371-9 (2023).
Eythorsson, E., Runolfsdottir, H. L., Ingvarsson, R. F., Sigurdsson, M. I. & Palsson, R. Rate of SARS-CoV-2 Reinfection During an Omicron Wave in Iceland. JAMA network open. 5 (2022).
Merad, M., Blish, C. A., Sallusto, F. & Iwasaki, A. The immunology and immunopathology of COVID-19. Science 375, 1122–1127. https://doi.org/10.1126/science.abm8108 (2022).
Sanaie, S. et al. Immune response variables and viral mutations impact on COVID-19 reinfection and relapse. Int. Immunopharmacol. 100, 108108–108108 (2021).
Yeoh, Y. K. et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 70, 698–706. https://doi.org/10.1136/gutjnl-2020-323020 (2021).
Kim, M. & Kim, C. H. Regulation of humoral immunity by gut microbial products. Gut Microbes. 8, 392–399. https://doi.org/10.1080/19490976.2017.1299311 (2017).
Pickard, J. M., Zeng, M. Y., Caruso, R. & Núñez, G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89. https://doi.org/10.1111/imr.12567 (2017).
Chu, H. & Mazmanian, S. K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 14, 668–675. https://doi.org/10.1038/ni.2635 (2013).
Bittinger, K. et al. Bacterial colonization reprograms the neonatal gut metabolome. Nat. Microbiol. 5, 838–847 (2020).
Hu, Z. et al. CoronaVac and BBIBP-CorV vaccines against SARS-CoV-2 during predominant circulation of Omicron BA.5.2 and BF.7 in China, a retrospective cohort study. J. Med. Virol. 95, e29143. https://doi.org/10.1002/jmv.29143 (2023).
Campbell, C. et al. Crosstalk between gut microbiota and host immunity: impact on inflammation and immunotherapy. Biomedicines 11 https://doi.org/10.3390/biomedicines11020294 (2023).
Oh, J. Z. et al. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 41, 478–492. https://doi.org/10.1016/j.immuni.2014.08.009 (2014).
Ng, S. C. et al. Gut microbiota composition is associated with SARS-CoV-2 vaccine immunogenicity and adverse events. Gut 71, 1106–1116. https://doi.org/10.1136/gutjnl-2021-326563 (2022).
Hagan, T. et al. Antibiotics-driven gut Microbiome Perturbation alters immunity to vaccines in humans. Cell 178, 1313–1328e1313. https://doi.org/10.1016/j.cell.2019.08.010 (2019).
Kim, D. et al. Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin. Nat. Med. 22, 524–530. https://doi.org/10.1038/nm.4075 (2016).
Lynn, D. J., Benson, S. C., Lynn, M. A. & Pulendran, B. Modulation of immune responses to vaccination by the microbiota: implications and potential mechanisms. Nat. Rev. Immunol. 22, 33–46. https://doi.org/10.1038/s41577-021-00554-7 (2022).
Ling, Z., Liu, X., Cheng, Y., Yan, X. & Wu, S. Gut microbiota and aging. Crit. Rev. Food Sci. Nutr. 62, 3509–3534. https://doi.org/10.1080/10408398.2020.1867054 (2022).
Chen, Y. et al. A comprehensive, longitudinal analysis of humoral responses specific to four recombinant antigens of SARS-CoV-2 in severe and non-severe COVID-19 patients. PLoS Pathog. 16, e1008796. https://doi.org/10.1371/journal.ppat.1008796 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics 31, 1674–1676. https://doi.org/10.1093/bioinformatics/btv033 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11 https://doi.org/10.1186/1471-2105-11-119 (2010).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. https://doi.org/10.1038/nbt.3988 (2017).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60. https://doi.org/10.1038/nmeth.3176 (2015).
Acknowledgements
This work was supported by grants from the Fundamental Research Funds for the Central Universities [grant number 2022ZFJH003], the Zhejiang Plan for the Special Support for Top-notch Talents [number 2022R52029], and Shandong Provincial Laboratory Project [grant number SYS202202].
Author information
Authors and Affiliations
Contributions
Jingying Pan: Conceptualization, Data curation, Formal analysis, Methodology, Software, Visualization, Writing an original draft. Xiaodi Zhang: Data curation, Formal analysis, Methodology. Danrong Shi: Data curation, Formal analysis, Methodology, Software. Xuebin Tian: Data curation, Investigation, Methodology. Lijun Xu: Data curation, Methodology. Xiangyun Lu: Data curation, Formal analysis, Methodology. Mingqing Dong: Data curation, Methodology. Peng Yao: Data curation, Methodology. Zhaoyi Pan: Data curation, Formal analysis, Methodology. Zongxin Ling: Supervision, Validation, Writing – review & editing. Nanping Wu: Supervision, Validation, Resources, Writing – review & editing. Hangping Yao: Conceptualization, Methodology, Project administration, Resources, Writing – review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, J., Zhang, X., Shi, D. et al. Short-chain fatty acids play a key role in antibody response to SARS-CoV-2 infection in people living with HIV. Sci Rep 14, 31211 (2024). https://doi.org/10.1038/s41598-024-82596-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-82596-0
