
Abstract
Colorectal cancer (CRC) is one of most common tumors worldwide, causing a prominent global health burden. Cell senescence is a complex physiological state, characterized by proliferation arrest. Here, we investigated the role of cellular senescence in the heterogeneity of CRC. Based on senescence-associated genes, CRC samples were classified into different senescence patterns with different survival, cancer-related biological processes and immune cell infiltrations. A senescence-related model was then developed to calculate the senescence-related score to comprehensively explore the heterogeneity of each CRC sample such as stromal activities, immunoreactivities and drug sensitivity. Single-cell analysis revealed there were different immune cell infiltrations between low and high senescence-related model genes enrichment groups, which was confirmed by multiplex immunofluorescence staining. Pseudotime analysis indicated model genes play a pivotal role in the evolution of B cells. Besides, intercellular communication modeled by NicheNet showed tumor cells with higher enrichment of senescence-related model genes highly expressed CXCL2/3 and CCL3/4, which attracted immunosuppressive cell infiltration and promoted tumor metastasis. Finally, top 6 hub genes were identified from senescence-related model genes by PPI analysis. And RT-qPCR revealed the expression differences of hub genes between normal and CRC cell lines, indicating to some extent the clinical practicability of senescence-related model. To sum up, our study explores the impact of cellular senescence on the prognosis, TME and treatment of CRC based on senescence patterns. This provides a new perspective for CRC treatment.
Similar content being viewed by others


Introduction
CRC, as a prominent global health burden, has posed a threat to human. CRC ranks third in terms of incidence with nearly 1.9 million new cases, following breast and lung cancer. But it is second in terms of mortality, with more than 935,000 deaths in 20201. Besides, the majority of CRC occur in individuals 50 years of age and older. At each stage of diagnosis, CRC patients younger than 50 years have a higher 5-year relative survival than older counterparts. And older patients are less likely than younger counterparts to receive any aggressive treatment, such as surgery, recommended adjuvant or neoadjuvant therapy2.
Cellular senescence is identified as a complicated stress response related with 4 interdependent hallmarks: secretory phenotype, deregulated metabolism, cell cycle arrest, as well as macromolecular damage3. Recent studies have shown that senescence contributes to several physiological processes such as embryonic development, regeneration, reprogramming, as well as wound healing4,5. However, the involvement of cellular senescence in various physiological and pathological processes can be explained not only by proliferation arrest, but even primarily by cell–cell interactions and secretion of factors affecting surrounding cells and compartments6. Due to epigenetic changes occurred in senescent cell, it may play a profound role in tissue homeostasis, organ function, cell regulation and secondary amplified immune network responses. These changes may contribute to create potential advantage for tumor growth7. Currently, several studies have reported some biomarkers of cellular senescence. For example, elevated lysosomal beta-galactosidase (encoded by GLB1) activity universally occurs in senescence cells, which has been exploited as a biomarker for cellular senescence, as known as senescence-associated beta-galactosidase (SA-beta-gal) activity8. Thus, investigating the effects of senescence in CRC may provide novel perspectives for further understanding of CRC heterogeneity.
In this study, we explored the expression of GLB1 in the Pan-cancer and found that it is high-expressed in CRC samples compared to normal tissue samples. Furthermore, immunohistochemistry (IHC) staining of anti-GLB1 performed on tissue microarray (TMA) from Ruijin hospital (RJH cohort) validated the above results. Based on human senescence-associated genes (SAGs) downloaded from CellAge Database, CRC samples were classified into different senescence patterns. We then investigated the differences in cancer-related hallmarks, immune and stromal activities among these senescence patterns. A scoring system, called senescence-related model, was developed to comprehensively elucidate the heterogeneity of CRC. Based on this model, senescence related score (SRS) of each CRC sample was calculated. Survival analysis indicated that SRS had the ability to distinguish CRC samples with different overall survive (OS) and progression free survival (PFS). Stromal activities such as epithelial-mesenchymal transition (EMT), angiogenesis, stromal score, TGF-β signaling and infiltration of immune suppressor cells, were more active in CRC samples from the high-risk group. On the contrary, CRC samples with low SRS have higher expression levels of immune biomarker genes and infiltration of immune effector cells. Single-cell analysis indicated senescence-related model genes played an important role in B cell evolution. And tumor cells highly expressed CXCL2/CXCL3, CCL3/CCL4 ligands, which might led to the recruitment of myeloid-derived suppressor cells (MDSC), mesenchymal stem cells (MSC), tumor-associated macrophages (TAM) and tumor-associated neutrophils (TAN) into tumor niches9. As expected, there also were significant differences in responses to chemotherapy and immunotherapy between low- and high-risk groups. Taken together, with multi-omics data we depicted the landscape and distinct patterns of cellular senescence in CRC, providing further insight of tumor heterogeneity and treatment strategy.
Materials and methods
Data requiring
The RNA-seq data of Pan-cancer, including 33 types of cancer, was downloaded from UCSC Xena (https://xena.ucsc.edu/). And The gene expression matrixes and clinical annotation of CRC and normal samples were downloaded from the Gene Expression Omnibus (GEO) database and The Cancer Genome Atlas (TCGA) database. In this study, 4 reliable CRC cohorts were collected for an in-depth analysis, including GSE39582, GSE103479, GSE17536 and TCGA-COADREAD. For microarray data from GEO database, a robust multi-array averaging method was utilized to perform background adjustment and quantile normalization based on the corresponding platforms annotation. During data processing, if multiple probes correspond to the same gene symbol, the average value is taken as the gene expression level. For the data of TCGA, the RNA-seq data of gene expression was downloaded from Genomic Data Commons (GDC, https://portal.gdc.cancer.gov/) in the value of fragments per kilobase of transcript per million fragments mapped reads (FPKM). Meanwhile, clinical annotation information for samples, such as survival, pathological stage, was also downloaded from the GEO and TCGA databases. In addition, we also collected the copy number variation (CNV) information and the somatic mutation data of CRC samples from the TCGA database.
To investigate the role of cell senescence in CRC, we downloaded 279 SAGs from CellAge Database in Human Ageing Genomic Resources (HAGR, https://genomics.senescence.info/). CellAge is a database of human senescence-related genes based on gene manipulation experiments and gene expression profiles. In order to reduce the batch effect, the RNA expression data from the GSE39582, GSE103479 and the TCGA cohorts were processed by “sva” R package. Samples with no OS information or an OS time of 0 were excluded from the study.
Preparation of single-cell suspension and droplet-based scRNA-seq
In this study, four tumor tissue samples were collected from patients who underwent colorectal cancer radical surgery with informed written consent at the Shanghai Minimally Invasive Surgery Center of Ruijin Hospital. And this study was approved by the Ethics Committee of Ruijin Hospital, Shanghai Jiao Tong University, School of Medicine.
After removing fat tissue and visible blood vessels, tissue samples were processed into single-cell suspensions by mechanical and enzymatic dissociation. By following the manufacturer’s protocol of 10X Chromium 3’ v3 kit (10X Genomics, Pleasanton, CA), single-cell suspensions were processed. Library was prepared and sequencing was performed on the NovaSeq 6000 platform (Illumina, Inc., San Diego, CA). In this study, we transformed raw sequencing reads into fastq file by the bcl2fastq2 Conversion Software (v2.20, Illumina) and conducted quality control by FastQC software (Version 0.11.9). Based on the human reference, GRCh38 (GENCODE v32/Ensembl 98), standard pipelines of cell ranger were used to do sequence processing.
Immunohistochemistry and multiplex immunofluorescence staining of tissue microarray
The tissue microarrays (TMA), containing 75 CRC with different clinical outcomes (such as AJCC-TNM stage) and paired adjacent normal paraffin-embedded specimens, were supplied by the Shanghai Minimally Invasive Surgery Center of Ruijin Hospital (Shanghai, China) following the guidelines by the Ethics Committee of Ruijin Hospital. All patients signed written and informed consent before the study. The IHC and mIF staining were performed using TMA slides as previously described10,11. The anti-GLB1 (1:500 dilution; ab305174) was for IHC staining. The anti-CD3 (1:500 dilution; ab237772), the anti-CD19 (1:5000 dilution; ab213363), the anti-PanCK (1:200 dilution; ab7753) were for mIF staining. And DAPI (dark blue) was used as a nuclear counter stain. Two pathologists, blinded to clinical information, analyzed the staining intensity of specimens using ImageJ software (National Institutes of Health, USA).
Differentially expressed SAGs screening and enrichment analysis
In this study, we combined GSE39582 and GSE103479 into the GEO cohort. Using “limma” R package, DeSAGs were screened between normal and CRC samples in the TCGA and GEO cohorts, respectively. And SAGs with FDR < 0.05 were considered as DeSAGs. Then, the overlapping DeSAGs between the TCGA and GEO cohorts were collected by “VennDiagram” R package for subsequent enrichment analysis. After converting gene symbol of the overlapping DeSAGs to Entrez Gene ID using “org.Hs.eg.db” R package, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of differential genes using “clusterProfiler” R package. When the q value was less than 0.05, the genes was considered to be enriched in this pathway.
Consensus clustering analysis of DeSAGs
Based on the expression of overlapping DeSAGs, “ConsensusClusterPlus” R package was used for consensus clustering analysis to divide CRC samples into different senescence patterns in pooled cohort (combination of TCGA and GEO cohorts). The appropriate number of clustering was determined by CDF curve. PCA was conducted to verify whether there were distinctions among these clusters. PCA is a mathematical algorithm for reducing the dimensionality of the data, improving interpretability while minimizing information loss12. To investigate whether there were significant differences of survival between clusters, “survival” R package was utilized to perform survival analysis and Kaplan–Meier (K–M) curves were plotted by “survminer” R package.
The differences of biological characteristics between clusters
To explore differences in biological processes of CRC samples between senescence patterns, GSVA, a non-parametric unsupervised method for estimating gene set enrichment variation13, was performed on RNA-seq data of CRC samples using “GSVA” R package. The hallmark gene set (h.all.v7.4.symbols) downloaded from Molecular Signatures Database (MSigDB) served as a reference gene set. Moreover, we utilized Tumor Immune Dysfunction and Exclusion (TIDE) website (http://tide.dfci.harvard.edu/) to calculated TIDE score of each CRC sample, which may serve as a surrogate biomarker to predict response to immune checkpoint inhibitors (ICIs)14. On this basis, this study further predicted the abundance of infiltrating 22 immune cell subsets in CRC samples by the CIBERSORT algorithm15.
Development of senescence-related model
In this study, we constructed a scoring system, called senescence-related model, to comprehensively quantify the senescence patterns of CRC samples. First, we utilized “limma” R package to screen for DEGs with absolute fold change (FC) greater than 2 and FDR less than 0.05 between different senescence patterns. We used the GEO cohort (GSE39582, GSE103479) as the training set, the TCGA cohort as the internal validation set, and GSE17536 as the external validation set. In the training set, these DEGs were then subjected to univariate Cox regression analysis for identifying DEGs associated with OS (p < 0.05), termed prognostic DEGs. Subsequently, prognostic DEGs were subjected to LASSO regression analysis for construction of senescence-related model using “glmnet” R package. After 10-Fold cross-validation, prognostic DEGs with non-zero coefficients were retained for model building. We utilized “RCircos” and “maftools” R packages to display CNV and mutation information of genes in model. Based on this model, SRS of each CRC sample was calculated and the specific method was as follows:
In this formula, Coef and Exp represent the coefficient and expression level of gene in this model, respectively. We evaluated the correlation between SRS and clinical information, including pathological stage, as well as AJCC-TNM stage. Based on median value of SRS, CRC samples in the training set were classified into low- and high-risk groups. Kaplan–Meier survival analysis was performed to investigate the differences of OS and PFS between low- and high-risk groups. The ROC curves at 1-, 3-, 5-year were plotted using “timeROC” R package to verify the value of SRS in prognostic prediction. The AUC can represent the prognostic prediction ability of SRS. The larger the AUC, the stronger the prediction ability. Same as above method, we validated the prognostic value of SRS in the internal and external validation sets, respectively. Apart from CRC, we also explored the role of SRS in pan-cancer prognosis. We downloaded the RNA-seq and survival data of pan-cancer samples from TCGA. According to the formula mentioned above, we calculated the SRS of pan-cancer samples, and then classified them into low and high-risk groups based on median value of SRS. Survival analysis was performed to reveal the differences in survival between low- and high-risk groups.
Construction of nomogram for survival prediction
In this study, we combined SRS of CRC samples with clinical information, including age, gender, pathological stage, as well as AJCC-TNM stage. Univariate and multivariate Cox regression analysis were conducted to determine the independent indicators related with survival. After analysis, indicators with p-value less than 0.05 were considered independent prognostic indicators. Then, these independent prognostic indicators were used to develop a nomogram for quantifying survival rate of each CRC sample. At the same time, ROC curves and calibration curves at 1-, 3- and 5-year were plotted to show the reliability and accuracy of nomogram using “survivalROC” and “rms” R packages. And, decision curve analysis (DCA) was performed to evaluate the net benefit of prognostic indicators and nomogram in CRC survival. DCA is a method that can evaluate the utility of models for decision making16.
Prediction of consensus molecular subgroups (CMSs)
CMS provides a comprehensive framework to capture the inherent heterogeneity of CRC through gene expression levels17. In this study, based on the RNA-seq of CRC samples, “CMScaller” R package was utilized to predict CMS of each CRC sample. And some known biological processes were compared between different CMSs. We then investigated the proportion of different CMSs in senescence patterns and SRS. In order to more intuitively show the relationship between SRS, senescence patterns, and CMS, alluvial figure was plotted using “ggalluvial” R package.
Exploration of TME between low- and high-risk groups
According to previous studies18,19,20, we collected 16 gene sets that was associated with some known biological processes, such as antigen processing machinery, immune checkpoint, EMT markers, pan-fibroblast TGFb response signature (Pan-FTBRS) and so on. Besides, we also obtained gene sets of the cancer-immunity cycle from TIP21. There are 7 critical steps in the cancer-immunity cycles, including Step1: release of cancer antigens; Step2: cancer antigen presentation; Step3: priming and activation of anticancer immune; Step4: trafficking of immune cells to tumor; Step5: infiltration of immune cells to tumor; Step6: recognition of cancer cell by T cells; Step7: killing of cancer cells22. We calculated the activities of these biological processes and steps in cancer-immunity cycles using “GSVA” R package. And the relevance of SRS to these activities were explored using “corrplot” R package. To further compare immune and stromal activation between CRC samples with different SRS, Estimation of STromal and Immune cells in MAlignant Tumours using Expression data (ESTIMATE) algorithm, a method that infers the fraction of stromal and immune cells in tumor samples using gene expression signatures23, was adopted to predict the immune and stromal score of CRC samples. Moreover, the expression levels of immune biomarker genes, cancer-associated biological enrichment conducted by Gene Set Enrichment Analysis (GSEA), and immune cells infiltration predicted by CIBERSORT algorithm were also compared between low- and high-risk groups.
Single-cell RNA sequencing (scRNA-seq) analysis
In the RJH cohort, the scRNA-seq of 4 CRC samples was used to explore senescence-related model genes. We first filtered and standardized the scRNA-seq data using “Seurat” R package. After standardization, the 1500 genes with the largest variance were reserved for subsequent analysis. PCA was then conducted to reduce dimensionality of data. And t-distributed stochastic neighbor embedding (t-SNE) method was applied to sort cells into different clusters. The cell annotation of each cluster was conducted by “SingleR” R package with reference of CellMarker24. In order to calculate the activity of senescence-related model genes in cells, we utilized to “AUCell” R package to calculated the AUC of each cell with the reference to model genes and then mapped the AUC to the corresponding cell. Cells that express more genes from senescence-related model will exhibit higher AUC values than cells expressing fewer genes. The evolutionary trajectory of cells and the role of model genes in cell evolution were predicted by “monocle” R package. Finally, “nichenetr” R package was utilized to infer the interactions between epithelial cells (tumor cells) and surrounding cells25. Genes which are expressed in larger than 10% cells of clusters were considered for ligand-receptor interactions. In paired ligand-receptor activity analysis, we extracted top 100 ligands and top 1000 targets of differential expressed genes of “sender cell” and “affected cell”, respectively. And we focused on investigating the regulation of epithelial cell on surrounding cells. The scRNA-seq of GSE132465 (including 23 CRC samples) was also analyzed to verify the reliability of the above results.
Exploring the role of SRS in drug treatment
Based on the study by Mini et al.26, CRC samples with disease recurrence within 3 years after chemotherapy were considered chemotherapy-resistant and CRC samples without disease recurrence within 5 years were considered chemotherapy-sensitive. And comparison of SRS between chemotherapy-resistant and chemotherapy-sensitive groups were conducted. To further investigate whether there were the differences in the response of CRC samples to chemotherapy between low- and high-risk groups, we utilized “pRRophetic” R package to predict the half maximal inhibitory concentration (IC50) of 5-fluorouracil (5-FU) of each CRC samples. IC50 is a quantitative measure that indicates how much of a specific inhibitory substance is required to inhibit. In terms of immunotherapy, TIDE (http://tide.dfci.harvard.edu/) and SubMap (https://cloud.genepattern.org/gp) algorithms were used to infer the potential response of CRC samples to immunotherapy. According to the annotation file subtypes data27, the response of CRC samples to anti-PD-1/CTLA4 immunotherapy was predicted and compared between low- and high-risk groups. GSE35640, downloaded from the GEO database, is a data set containing gene expression data and information on response to immunotherapy in melanoma tumor samples28. According to senescence-related model, we calculated SRS of each sample, then compared the differences of SRS between responders and non-responders.
Prediction of potential drug for CRC
Based on previous study29, the somatic mutation data and expression profile data of human cancer cell lines (CCLs) were downloaded from the Broad Institute Cancer Cell Line Encyclopedia (CCLE) project30. Then, the sensitivity data for 1448 compounds to 482 CCLs was acquired from the PRISM Repurposing dataset (PRISM; https://depmap.org/portal/prism/) and the sensitivity data for 481 compounds to 835 CCLs was acquired from Cancer Therapeutics Response Portal (CTRP; https://portals.broadinstitute.org/ctrp). The area under the dose–response curve (AUC) was calculated to represent the sensitivity of the drug. The lower AUC indicates the higher sensitivity to drug. In the prediction of drug sensitivity, drugs/compounds that lack of more than 20% data were excluded and K-nearest neighbor (k-NN) imputation was performed to identify and replace missing values for AUC.
The protein–protein interaction network (PPI) of genes in model
In order to identify the interaction network between genes from senescence-related model, we first performed PPI analysis on these genes via STRING (https://string-db.org/), a database that contributes to explore the interaction of genes and further unearth hub genes. The active interaction sources included text-mining, experiments, databases, co‑expression, neighborhood, gene fusion, co‑occurrence. They were represented by different color lines such as yellow, purple, light blue, black, green, red and blue. And high confidence (0.700) was selected as minimum required interaction score. Then, cytoHubba, a plug-in of Cytoscape (Version: 3.9.1), was utilized to identify the hub genes form PPI. In addition, we further investigated the association between the expression levels of hub genes and the infiltration of 22 immune cells.
Cell lines culture and quantitative real-time polymerase chain reaction (RT-qPCR)
In this study, we validated the expression levels of hub genes between normal (NCM-460) and CRC cell lines (HCT116, HT-29). All cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and stored in the Shanghai Institute of Digestive Surgery. Cells were cultured in McCoy's 5A or RPMI-1640 (Gibco, Grand Island, NY, USA) supplemented with 1% penicillin/streptomycin (Gibco) and 10% fetal bovine serum (FBS) at 37 °C with 5% CO2.
The total RNA of cell lines was extracted using FastPure® Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, China). The NanoDrop 2000 spectrophotometer (Thermo) then was used to quantify RNA. After reverse transcription of RNA to cDNA by HiScript® RT SuperMix for qPCR with gDNA wiper (Vazyme, China), we performed RT-qPCR on cDNA using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China). And the cycler protocol is as follows: Stage 1: 30 s at 95 °C, Stage 2: 40 cycles of 10 s at 95 °C, 30 s at 60 °C, and Stage 3: 15 s at 95 °C, 60 s at 60 °C, 15 s at 95 °C. GAPDH was exploited as an internal reference. The mRNA relative expression levels of hub genes were calculated by 2−ΔΔCt methods. The primer sequences used for analysis are listed in Table S1.
Statistical analysis
The Wilcoxon rank-sum test was used to compare differences between two groups, and the Kruskal–Wallis test was used in the comparison of three or more groups. The Kaplan–Meier method was applied to draw K–M curves showing differences in survival between high- and low-risk groups. Independent prognostic indicators of CRC were determined by univariate and multivariate Cox regression analysis. ROC curves were drawn to assess prognostic values for indicators and nomogram. All statistical analyses were performed in R software (version 4.2.0). And all methods were performed in accordance with the relevant guidelines and regulations.
Ethical approval
The Ethics Committee of Ruijin Hospital, Shanghai Jiao Tong University, School of Medicine approved this research.
Informed consent
The informed consent of all patients was obtained before the study.
Results
Exploring the expression levels of SA-beta-gal
In the Pan-cancer cohort, we found the expression levels of GLB1 in rectum adenocarcinoma (READ) and colon adenocarcinoma (COAD) ranked 3rd and 5th among 33 cancers, respectively (Fig. 1a). Besides, compared with normal tissue, the expression levels of GLB1 were higher in READ and COAD (Fig. 1b). For further validation of GLB1 expression in CRC, we performed IHC staining for GLB1 in TMA from the RJH cohort. The representative IHC photographs of normal and tumor tissue were displayed in Fig. 1c, d. The quantitative result showed that tumor tissue had significantly higher GLB1 expression than that in normal tissue (Fig. 1e). These results indicated that cell senescence (GLB1 encoding SA-beta-gal, a biomarker of cell senescence) may have a pivotal impact on CRC.

Exploring the expression of GLB1 encoding senescence-associated beta-galactosidase in Pan-cancer and the RJH cohort. (a) The expression levels of GLB1 in Pan-cancer. (b) Differences in GLB1expression levels between normal and tumor samples in Pan-cancer. (c) Representative IHC photographs of GLB1 in normal patient from the RJH cohort. Left: magnification: ×40; Right: magnification: ×200. (d) Representative IHC photographs of GLB1 in CRC patient from the RJH cohort. Left: magnification: ×40; Right: magnification: ×200. (e) Comparison of immunohistochemical staining intensity of GLB1 between normal and CRC patients from the RJH cohort.
Enrichment analysis of differentially expressed SAGs (DeSAGs)
Based on 256 SAGs after data normalization, differential expression analysis was conducted to identify 204 DeSAGs and 184 DeSAGs between normal and tumor tissue samples in TCGA and GEO cohorts, respectively. Figure S1a, b showed top 20 DeSAGs with absolute maximal FC in the form of heatmap in the TCGA and GEO cohorts, respectively. 155 overlapping DeSAGs from two cohorts were then collected for subsequent enrichment analysis (Fig. S1c). GO enrichment analysis revealed that these DeSAGs were enriched in pathways related with aging, cell aging, cell senescence both in TCGA and GEO cohorts (Fig. S1d, e). Similarly, KEGG enrichment analysis showed these DeSAGs were enriched in senescence pathways, in addition to CRC, immunity, and infection (Fig. S1f, g). These results indicate that DeSAGs may have an impact on TME characterization of CRC.
Consensus clustering of senescence patterns
To clarify the impact of DeSAGs on CRC, 1226 CRC samples in the pooled cohort were classified into 4 clusters with different senescence patterns based on DeSAGs. There were 451 CRC samples in cluster1, 451 CRC samples in cluster2, 193 CRC samples in cluster3, 131 CRC samples in cluster4, respectively (Fig. 2a). The cumulative distribution function (CDF) curves for k = 2–9 were plotted, in which k represented the number of clusters (Fig. S2a). And the relative change in area under CDF curves were demonstrated in Fig. S2b. The result of principal component analysis (PCA) showed there were boundaries between CRC samples from different clusters, suggesting CRC samples with different senescence patterns might have significantly different characterizations (Fig. 2b). Survival analysis revealed significant differences in OS between 4 clusters, with CRC samples in cluster3 having the worst OS (p < 0.001; Fig. 2c). Similar to OS results, K-M curves showed that CRC samples in cluster3 also had the worst PFS with p < 0.001 (Fig. S2c).

Consensus clustering for different senescence patterns. (a) Consensus clustering of CRC samples based on DeSAGs. (b) The result of PCA on CRC samples with different senescence patterns. (c) The overall survival (OS) comparison between CRC samples from different clusters. (d) The comparison of cancer-related hallmark between CRC samples from different clusters. (e) The comparison of TIDE score between CRC samples from different clusters. (f) Differences in immune cell infiltration between clusters predicted by the CIBERSORT algorithm. *: p < 0.05; **: p < 0.01; ***: p < 0.001. (g) Differences in biological processes between clusters predicted by GSVA. *: p < 0.05; **: p < 0.01; ***: p < 0.001.
In this study, we further explored the differences in biological processes, immune cells infiltration and stromal characteristics between 4 clusters. The result of GSVA showed that CRC samples from different clusters had notable distinctions in cancer-related biological processes (Fig. 2d). For example, the processes of inflammatory response, EMT, angiogenesis and TGF-β signaling in cluster3 were more active compared to other clusters. In the comparison of Tumor Immune Dysfunction and Exclusion (TIDE) score, CRC samples from cluster3 had higher TIDE score than other CRC samples with p < 2.2e−16 (Fig. 2e), indicating CRC samples in cluster3 may have tumor immune dysfunction and rejection. CIBERSORT algorithm was used to predict the infiltration of immune cells in TME of CRC samples. As shown in Fig. 2f, the infiltration of immune effector cells, such as CD8+ T cell, plasma cell, as well as activated NK cell, was lower in the CRC samples in cluster3 than that in other clusters. Inversely, macrophages (including M0, M1, M2) were highly infiltrated in TME of CRC samples from cluster3. Finally, some known biological processes were quantified and compared between different clusters (Fig. 2g). Although antigen processing machinery, CD8+ T effector were more active in CRC samples from cluster3, the level of stromal activities such as Pan-F-TBRS, angiogenesis, EMT 2/3 were also higher in CRC samples from cluster3. The above findings demonstrated CRC samples with different senescence patterns had significant differences in prognosis, biological activity, and TME, providing a new avenue to understand the heterogeneity of CRC.
Construction of senescence-related model
A total of 611 differently expressed genes (DEGs) were identified from 4 clusters with different senescence patterns (Fig. S3a). In the training set, univariate Cox regression analysis identified 117 prognostic DEGs (p < 0.05) for further model construction (Fig. S3b). The hazard ratio (HR) of each prognostic DEG was calculated, HR < 1 meant protective factor for prognosis, and HR > 1 meant risk factor for prognosis. The coefficients of prognostic DEGs were calculated by LASSO regression analysis (Fig. S3c). And 10-Fold cross-validation was performed to determine the 23 prognostic DEGs for construction of senescence-related model (Fig. S3d). The CNV information and position of model genes were displayed in Fig. S3e. In terms of mutation, DUSP27 was the gene with the highest mutation frequency, and missense mutation was the most common type of mutation (Fig. S3f).
Clinical implications of senescence-related model
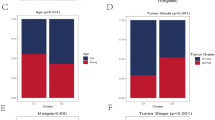
According to the coefficients and expression level of model genes, SRS of CRC samples were calculated. We could find that CRC samples in higher AJCC-TNM stage had higher SRS (Fig. 3a–c). Meanwhile, CRC samples in pathologically advanced stage also had higher SRS than that in early stage (Fig. 3d). These results suggested that there may be a strong association between SRS and survival of CRC. Therefore, we further investigated the difference in prognosis among CRC samples with different SRS. In the training set, CRC samples with high SRS had poorer OS than that with low SRS (p < 0.001; Fig. 3e). Similar outcomes were observed in the internal and external validation sets (Fig. 3f-g). In the training set, the area under receiver operating characteristic (ROC) curves of SRS in predicting 1-, 3-, 5-year OS was 0.710, 0.706 and 0.691, respectively (Fig. 3h). And Fig. 3i-j displayed the AUC of 1-, 3-, 5-year OS prediction in the internal and external validation sets. In addition to OS, we also investigated the role of SRS in predicting PFS of CRC samples. As shown in Fig. S4a–c, there were significant differences of PFS between low- and high- risk groups in the training, internal validation and external validation sets. Figure S4d, f showed the ROC curves of SRS in predicting 1-year, 3-year and 5-year PFS of CRC samples in the training, internal validation and external validation sets. Based on the above results, we can infer that SRS can be used as a reliable and effective auxiliary tool for the prognosis prediction of CRC samples. Moreover, mutation landscapes of low- and high-risk groups were displayed in Fig. 3k, l. In pan-cancer, SRS still had the ability to distinguish the prognosis of different samples (Fig. S5).

Clinical relevance of senescence-related model. (a–d) The comparison of SRS between CRC samples in AJCC-T stages, AJCC-N stages, AJCC-M stages, as well as pathological stages, respectively. And p < 0.05 is considered statistically significant. (e–g) Survival analysis between CRC samples from low- and high-risk groups in the training set, internal and external validation sets. And p < 0.05 is considered statistically significant. (h–j) ROC curves of SRS in predicting OS at 1-, 3-, 5-year in the training set, internal and external validation sets, respectively. (k) The mutation landscape of genes in CRC samples from the low-risk group. Top 20 genes with highest mutation frequency displayed in the waterfall plotting. (l) The mutation landscape of genes in CRC samples from the high-risk group. Top 20 genes with highest mutation frequency displayed in the waterfall plotting.
Development of nomogram for survival prediction
After univariate/multivariate Cox regression analysis, we found that age, pathological stage, T stage, M stage and SRS were independent prognostic indicators (Fig. 4a, b). Based on these independent prognostic indicators, a nomogram for quantifying the 1-year, 3-year, and 5-year survival probabilities of CRC samples was developed (Fig. 4c). Compared with single prognostic indicator, the AUC of nomogram was higher, indicating that nomogram had better specificity and sensitivity in predicting prognosis of CRC samples (Fig. 4d). The results of DCA demonstrated net benefit of age, pathological stage, AJCC-T/M stage, SRS and nomogram within a range of clinically reasonable risk thresholds (Fig. 4e). The net reduction curves showed the number of reduced patients after the interventions based on the decision derived from age, pathological stage, AJCC-T/M stage, SRS and nomogram (Fig. 4f). By comparison, we found that the nomogram had the greatest net benefit and net reduction, indicating nomogram is superior to other indicators in making better clinical decisions. Finally, calibration curves at 1-, 3- and 5-year were drawn to illustrate the difference between nomogram-predicted and actual survival. As shown in Fig. 4g–i, the nomogram-predicted survival is basically consistent with the actual survival. Therefore, nomogram may be a complementary tool to better assess the prognosis of CRC. In addition, this further indicated the importance of SRS in tumor prognosis, inspiring us to explore the mechanism of CRC from the perspective of cellular senescence.

A nomogram for predicting CRC prognosis. (a) The forest plot of univariate Cox regression analysis of SRS and clinical indicators. (b) The forest plot of multivariate Cox regression analysis of SRS and clinical indicators. (c) A nomogram containing independent prognostic indicators, including age, AJCC-T stage, AJCC-M stage, pathological stage and SRS. (d) ROC curves of independent prognostic indicators. Higher AUC is more valuable in predicting prognosis. (e) Decision curve analysis (DCA) curves show net benefits of age, pathological stage, AJCC-T stage, AJCC-M stage, SRS and nomogram at a range of clinically reasonable risk thresholds. (f) The net reduction analysis indicates how many patient deaths could be prevented after the intervention, as determined by age, pathological stage, AJCC-T stage, AJCC-M stage, SRS and nomogram. (g–i) Calibration curves of nomogram at 1-, 3- and 5-year.
Predicting consensus molecular subgroups (CMSs) of CRC samples
To explore the relationship between senescence patterns, SRS and tumor characterization, we predicted CMS of each CRC sample. As demonstrated in Fig. S6a, 171 CRC samples were classified into CMS1; 312 CRC samples were classified into CMS2; 172 CRC samples were classified into CMS3; 311 CRC samples were classified into CMS4. There were significant differences of biological processes between 4 CMSs. For example, microsatellite instability (MSI) of CRC samples in CMS1 were higher than that of other CMSs. Oppositely, CRC samples in CMS2 had feature of microsatellite stability (MSS). Compared with other CMSs, the activities of differentiation, glycolysis, fatty acids were more active in CMS3. And CRC samples in CMS4 were characterized by TGF-β signaling, as well as EMT. This study further investigated the proportion of CMSs in each senescence patterns. As expected, CMS4 had the most prominent proportion in CRC samples from cluster3, which was consistent with the result of GSVA (Fig. S6b). At the same time, we found CMS4 was the largest proportion in the high-risk group (Fig. S6c). The alluvial figure demonstrated the relationship between SRS, CMSs and senescence patterns (Fig. S6d). And the specific number of low- and high-risk CRC samples in each CMS was shown in Fig. S6e. These results suggested significant heterogeneity in CRC, such as immune activity, metabolism status and tumor pathway activity. However, senescence patterns can distinguish CRC with different characteristics. This provides a new reference for the precision treatment of CRC.
Characterization of TME between CRC samples with different SRS
To investigate the effect of SRS on CRC TME, we explored the relevance of SRS to cancer-related biological processes and 7 critical steps in the cancer-immunity cycles. SRS was negatively related with CD8+ T effector, antigen processing machinery and immune checkpoint. Inversely, SRS was positively related with Pan-F-TBRS, as well as angiogenesis (Fig. 5a). In terms of cancer-immunity cycles, SRS was negatively related with all steps (Fig. 5b). These results suggested CRC samples with higher SRS may have an anti-tumor immune deficiency. Although there was no significant association between immune score and SRS (r = − 9.19e−03, p = 0.75; Fig. 5c), there was a positive correlation with stromal score (r = 0.37, p = 7.63e−41; Fig. 5d). Subsequently, the result of comparison of immune biomarker genes between low- and high-risk groups showed CRC samples with low SRS had higher expression levels of immune genes than that with high SRS (Fig. 5e). And GSEA further validated the similar results, CRC samples in low-risk group was enriched in immune-related pathways, including interferon alpha response and interferon gamma response (Fig. 5f). As for CRC samples in high-risk group, they were characterized by EMT, hypoxia, TGF-β signaling (Fig. 5g). The infiltration of 22 immune cells also illustrated immune effector cells, such as plasma cell, CD8+ T cell, activated NK cell were highly infiltrated in TME of CRC samples with low SRS. Inversely, the abundance of macrophage M0 and macrophage M2 in high-risk group were significantly higher than that in low-risk group (Fig. 5h). Hence, we inferred senescence-related model genes played an important role in immune cells infiltration in CRC TME, which in turn affected the response of CRC to immunotherapy.

The heterogeneity of CRC samples with different SRS. (a) The correlation of SRS with several known biological processes. Orange represents the positive correlation and blue represents the negative correlation. (b) The correlation of SRS with 7 critical steps in the cancer-immunity cycles. Orange represents the positive correlation and blue represents the negative correlation. (c) The relationship between SRS and immune score calculated by ESTIMATE algorithm. (d) The relationship between SRS and stromal score calculated by ESTIMATE algorithm. (e) Comparison of immune biomarker gene expression levels between low- and high-risk groups. (f) Enrichment of cancer-related hallmarks in CRC samples from the low-risk group. (g) Enrichment of cancer-related hallmarks in CRC samples from the high-risk group. (h) The differences in the infiltration of immune cells between the low- and high-risk groups.
Investigation of senescence-related model at the single-cell level
In the RJH cohort, we first performed quality control on scRNA-seq of 4 CRC samples. Cells with RNA number > 50 and mitochondrial RNA percentage < 10% were retained for subsequent analysis (Fig. S7a). 1500 genes with the largest variances were selected for PCA analysis (Fig. S7b). PCA analysis was conducted on these genes and top 20 PCs were selected for further dimensional reduction (Fig. S7c, d). Finally, cells were divided into 18 clusters (0–17 clusters) by t-SNE (Fig. S7e). all of these cells were annotated as T cell, B cell, epithelial cell, monocyte, neutrophils, NK cell, endothelial cell, and smooth muscle cell (Fig. 6a). AUC score of each cell was then calculated and projected onto the t-SNE diagram (Fig. 6b). AUCell scoring algorithm was applied to assign all cells into low- and high-AUC groups. We found two peaks in the AUC score of the cells. When the AUC score threshold was set to 0.062, 6098 cells showed relatively higher AUC score (Fig. 6c). Compared with high-AUC group, the proportion of B cells in the low-AUC group was higher, while the proportion of epithelial cells was opposite (Fig. 6d). To understand the role of model genes on B cells, we further performed analysis of scRNA-seq of B cells (Fig. S7f, g). The evolutionary trajectory of B cells with different clusters and states over pseudotime was displayed in Fig. S7h–j. As shown in Fig. 6e, B cells were annotated as 4 subsets, including plasma cell, memory B cell, naïve B cell, and geminal center B cell. The density of 4 B cell subsets over pseudotime was displayed in Fig. 6f. At the beginning of evolution, germinal center B cell appeared, followed by memory B cell and naïve B cell. Finally, plasma cells evolved. The expression level of each model gene over pseudotime was displayed in Fig. 6g. The expression levels of model genes in different B cell states and subsets were shown in Fig. S8. The evolutionary trajectory demonstrated germinal center B cell had two distinct evolutionary directions. One direction evolved into memory cells, and the other direction evolved into plasma cells (Fig. 6h). As expected, the expression levels of senescence-related model genes in different cells states were significantly different, indicating model genes may play an important role in evolution of B cells (Fig. 6i). Exploring cell–cell communication found that epithelial cell highly expressed CXCL2, CXCL3, while EGR1, FOS, IER3, DUSP1, JUNB and NFKBIA were predicted as potential target genes (Fig. 6j). The ligand-receptor pairs with the top potential interactions were also predicted (Fig. 6k).

Exploration of senescence-related model in scRNA-seq of the Ruijin hospital cohort (RJH cohort). (a) Cell annotation of scRNA-seq from the Ruijin cohort. (b) The t-SNE plots based on the AUC score of cells. Cell subsets with high AUC score are highlighted. (c) AUC scoring of senescence-related model genes. The threshold was determined as 0.062 and the 6098 cells exceeded the threshold value. (d) The comparison of cell subset proportions between low and high AUC score groups. (e) Cell annotation of B cell subsets. (f) The density changes of B cell subsets over pseudotime. (g) The heatmap shows the expression level of model genes in B cells over pseudotime. (h) The evolutionary trajectory along the B cell subsets, which is started from germinal center B cell. (i) The role of model genes in evolution of B cell subsets. (j) Top-correlated ligand activities inferred in epithelial cells (tumor cells) according to NicheNet. Heatmap showing regulatory potential of top-correlated ranked ligands in epithelial cells (tumor cells) and the downstream target genes in surrounding cells. (k) The heatmap showing potential interaction of ligand-receptor pairs between epithelial cells (tumor cells) and surrounding cells.
In GSE132465, cells from 23 CRC samples were classified into epithelial cell, stromal cell, T cell, B cell, mast cell, as well as myeloid cell (Fig. 7a). When the AUC score threshold was set to 0.054, 39,823 cells showed relatively higher AUC score (Fig. 7b). As shown in t-SNE plot, high-AUC score cells were mainly consisted of epithelial cell, stromal cell and myeloid cell (Fig. 7c), which indicated that CRC with high expression of model genes may be associated with activation of stromal and immune-suppressive activities. To this end, we investigated the cell–cell communication mechanisms of epithelial cell and surrounding cells according to the expression and downstream targets of ligand-receptor pairs. We found that epithelial cell showed high CCL3, CCL4 and ALCAM ligand activity (Fig. 7d). And the expressions of ligands in epithelial cell were shown in Fig. 7e. In addition, CCL3 and CCL4 encoding protein bound to receptor encoded by CCR1, whereas ligand encoded by ALCAM interacted with receptor encoded by CD6 (Fig. 7f), resulting in the expression of target genes such as CDKN1A, MMP9, and TGFB1 (Fig. 7g). These results indicated senescence-related model genes may play a non-negligible role in evolution of B cells. Meanwhile, epithelial cell developed interaction networks with surrounding cells, which potentially induced cellular senescence and promoted the remodeling of the TME. This also partly explained the difference in sensitivity of CRC to drug therapy, including chemotherapy and immunotherapy.

Validation of senescence-related model from a single-cell perspective. (a) Cell annotation of scRNA-seq from GSE132465. (b) AUC scoring of senescence-related model genes. The threshold was determined as 0.054 and the 39,823 cells exceeded the threshold value. (c) The t-SNE plots based on the AUC score of cells. Cell subsets with high AUC score are highlighted. (d) Top-correlated ligand activities inferred in epithelial cells (tumor cells) according to NicheNet. (e) Dot plots showing the intensity (dot intensity) and the expression percentage (dot size) of top- correlated ligands in epithelial cells (tumor cells). (f) Ligand-receptor pairs displaying potential interactions between epithelial cells (tumor cells) and surrounding cells. (g) Heatmap showing regulatory potential of top-correlated ranked ligands in epithelial cells (tumor cells) and the downstream target genes in surrounding cells.
Prediction of response to drug treatment in CRC samples with different SRS
Based on the positive correlation between SRS and AJCC-TNM stage and the effect of model genes on immune cells, this study further conducted mIF on CRC tissues of patients with different pathological stages. As shown in Fig. 8a, the infiltration of T cell (CD3) and B cell (CD19) in early CRC was higher than that in advanced stage. In addition, in the GEO cohort, CRC samples in high-risk group were more sensitive to anti-CTLA4 ICIs compared with that in low-risk group. On the contrary, CRC samples in low-risk group were more sensitive to anti-PD-1 ICIs compared with that in high-risk group (Fig. 8b). These results were validated in the TCGA cohort (Fig. 8c). To further validate the reliability of SRS in predicting response to immunotherapy, we compared the difference of SRS between non-responder and responder in the GSE3564, which contained immunotherapy information of melanoma patients. As expected, SRS of non-responders was significantly higher than that of responders (p = 0.033; Fig. 8d). The proportion of non-responders in high-SRS group was higher than that in low-SRS group (Fig. 8e). These results indicated CRC patients with low SRS were more likely to benefit from immunotherapy.

Responses of CRC samples with different SRS to drug therapy. (a) Multiplex immunofluorescence staining of CRC tissues in early stage and advanced stage. Yellow: CD3, far-red: CD19, red: PanCK, dark blue: DAPI. (b) The comparison of responses to anti-PD-1/ CTLA4 immunotherapy between low- and high-risk groups in the GEO cohort. (c) The comparison of responses to anti-PD-1/CTLA4 immunotherapy between low- and high-risk groups in the TCGA cohort. (d) The comparison of SRS between non-responders and responders. (e) The proportion of non-responders and responders in low- and high-SRS groups. (f) The comparison of SRS between CRC samples from chemotherapy-resistant and chemotherapy-sensitive groups. (g) The comparison of estimated IC50 of 5-FU between CRC samples from the low- and high-risk groups. (h) Top 6 potential compounds/drugs predicted to be positively or negatively associated with SRS in the PRISM database. (i) Top 6 potential compounds/drugs predicted to be positively or negatively associated with SRS in the CTRP database.
In the GEO cohort, CRC samples with chemotherapy were classified into chemotherapy-resistant and chemotherapy-sensitive groups. As shown in Fig. 8f, SRS of CRC samples in chemotherapy-resistant group were higher compared with that in chemotherapy-sensitive group (p < 0.001). In the prediction of response to chemotherapy, IC50 of 5-FU in high-risk group was also higher than that in low-risk group (p < 0.001; Fig. 8g). These results suggested that CRC samples with high SRS may be difficult to benefit from chemotherapy.
In order to complement drug treatment strategies of CRC, several compounds/drugs were predicted as potential treatment strategies for CRC samples with different SRS. Figure 8h showed top 6 compounds/drugs positively and negatively associated with SRS in the PRISM database. Similarly, we also predicted top 6 compounds/drugs positively and negatively associated with SRS in the CTRP database (Fig. 8i). Interestingly, we found the AUC of floxuridine (which can be rapidly bio-catabolized to 5-fluorouracil) and fluorouracil were positively related with SRS, suggesting that CRC patients with low SRS were more sensitive to fluorouracil. It further validated the result of Fig. 8g. All of these results indicated the potential value of SRS in the precision treatment of CRC and provided new potential therapeutic drugs for CRC.
Validation of hub genes in senescence-related model
This study performed protein–protein interaction network (PPI) analysis on 23 genes from senescence-related model, and network of these genes was displayed in Fig. 9a. Through cytoHubba, 6 genes were identified as hub genes, including CXCL13, MMP12, CXCL1, CXCL9, IL7R, and MMP3 (Fig. 9b). The relationship between the expression levels of hug genes and infiltrations of 22 immune cells was demonstrated in Fig. 9c.

PPI network of genes in senescence-related model. (a) PPI network of genes in model. Red line—indicates the presence of fusion evidence; Green line: neighborhood evidence; Blue line: cooccurrence evidence; Purple line: experimental evidence; Yellow line: text-mining evidence; Light blue line: database evidence; Black line: co-expression evidence. (b) Hub genes identified from genes in model. (c) Relationship between hub gene expression levels and immune cell infiltration. (d–i) Validation of expression levels of hub genes between normal cell line (NCM-460) and CRC cell lines (HCT 116, HT-29) by RT-qPCR. (d) CXCL13. (e) MMP12. (f) CXCL1. (g) CXCL9. (h) IL7R. (i) MMP3.
In order to preliminarily validate the clinical practicality of senescence-related model, we compared the relative expression levels of these hub genes between normal (NCM-460) and CRC cell lines (HCT 116, HT-29) by RT-qPCR. Figure 9d–i showed the relative expression levels of CXCL13, MMP12, CXCL1, CXCL9, IL7R, as well as MMP3, respectively. As we can see, all hub genes are underexpressed both in HCT 116 and HT-29 compared to NCM-460, which is consistent with HR of hub genes in CRC prognosis calculated by univariate Cox regression analysis.
Discussion
Cellular senescence, as a stable cell cycle arrest, plays a non-negligible role in the limitation of proliferative lifespan of diploid cells. There are different types of senescence in the human body. For example, replicative senescence is characterized by gradual shortening telomerases with each cell division, which is a biological response to prevent genomic instability. The downside of this biological response is the accumulation of damaged DNA31. With age, replicative senescence in cells may cause some age-related diseases, such as osteoarthritis32 and atherosclerosis33. Moreover, an accelerated senescence, as known as premature senescence, is different from replicative senescence. The main causes of premature senescence are some specific stimuli generated within the cells, including metabolic shock, genotoxic stress, and so on31,34,35. Several studies have revealed that carcinogenic stress, including overexpression of oncogenes and loss of tumor suppressor genes (TSGs), can also induce senescence36,37. When senescence occurs in tumors, it can inhibit the development and progression of tumor. Therefore, senescence is a physiological mechanism of tumor suppression, which can inhibit the progression of benign tumor lesions to malignant tumors38. Due to its antiproliferative effects, senescence may act as a potential mechanism of anti-cancer. At present, some researches by promoting cellular senescence have provided a new direction for anti-cancer therapy, which is called pro-senescence therapy39.
Senescent tumor cells can secrete a plethora of senescence-associated secretory phenotype (SASP), including inflammatory cytokines, immune modulators, proteases, chemokines, as well as growth factors, via paracrine mechanisms, resulting in non-negligible effects on the TME39,40. SASP has been considered a double-edged sword, as it can act on neighboring cells as well as on the recruitment and activation of immune cells, resulting in dual anti-tumor and pro-tumor effects41. For instance, the induction of senescence plays an important tumor suppressor role in early hepatocellular carcinoma. However, SASP secreted by senescence cells can suppress immune surveillance, favoring tumor progression in the advanced stages of the disease42. In addition, senescence also has a direct impact on immune cells, known as immunosenescence. By causing a battery of changes in the development and function of immune cells, immunosenescence can promote body’s susceptibility to disease38. Therefore, we infer that cellular senescence may play an important role in CRC development and progression.
This study provided a novel perspective on cell senescence for CRC research. CRC samples were classified into 4 clusters with different senescence patterns by SAGs from CellAge Database. There were significant differences of cancer related hallmarks between different clusters, such as inflammatory response, interferon-γ response, TNF-α signaling via NF-κB, TGF-β signaling. Several papers have reported that IFN-γ and TNF-α, as typical inflammatory cytokines, that can induce premature senescence in CD8+ T lymphocytes by activating p38MAPK and downregulating telomerase expression43,44. In addition to this, TGF-β signaling is involving in the immunosuppression in the TME, which can affect tumorigenesis through MDSC. Due to this suppressive activity, CD8+ T cells will be tolerated thus lose their tumor-killing effect45. As expected, we found that immune suppressor cells were highly infiltrated in TME of CRC samples with these highly active hallmarks (Cluster3). In contrast, immune effector cells were low-infiltrated in TME of CRC samples from Cluster3 compared to that from other clusters. Moreover, some stromal activities were significantly different between different clusters, such as angiogenesis, Pan-F-TBRS. This suggested that there was heterogeneity in CRC between different senescence patterns.
In order to comprehensively quantify the characteristics of each CRC sample, 23 DEGs were screened from different senescence patterns to construct a senescence-related model. Based on this model, SRS of all CRC was calculated for subsequent analysis. Because of positive correlation between SRS and clinical stage, we inferred SRS may act as a potential biomarker for prognosis prediction in CRC. Survival analysis confirmed it and SRS was further validated as an independent prognostic indicator by univariate/multivariate Cox regression analysis. Combined with other clinical prognostic indicators, a nomogram was constructed for predicting survival probability of each CRC sample. ROC curves and calibration curves illustrated nomogram had high accuracy and reliability in predicting prognosis. Therefore, SRS may be used as an auxiliary indicator for prognosis prediction of clinical CRC samples.
At the single-cell level, we found tumor cells, neutrophils, monocyte, and stromal cells had higher AUC score, while B cell had significantly lower AUC score. It indicated cellular senescence may affected the infiltration of immune cells and stromal cells in TME of CRC. Hence, we further investigated the role of senescence-related model genes in B cell evolution. In the evolution trajectory, model genes such as CXCL1, CXCL9, MMP3, MMP12 were highly expressed in naïve B cell and memory B cell. Meanwhile, model genes such as TMEM160, EGR3, PJA2, SLC2A3 were expressed at extremely low levels in plasma cells, suggesting these model genes may promote the evolution of B cells into naïve B cells and memory B cells rather than plasma cells. This partly explains why the infiltration of plasma cells was higher in the low-risk group. Therefore, we speculate that cellular senescence may have a profound impact on TME remodeling and heterogeneity formation in CRC. In order to verify our conjecture, we explored the communication of tumor cells with surrounding cells by ligand-receptor interactions. We found tumor cells expressed high level of CXCL2 and CXCL3. Previous study has revealed that CXCL2, as CXCR2 ligand, controlled neutrophil senescence in a cell-autonomous manner46. In lung metastasis of CRC, tumor-derived CXCL2 attracted M2 macrophage to infiltrate and promote lung metastasis47. Similarly, up-regulation of CXCL3 by activation of TGFβ signaling pathway could drive the aggregation of immunosuppressive neutrophils, promoting CRC liver metastasis48. In addition, the CCL3/CCL4-CCR1 ligand-receptor pair also affected the communication between tumor cells and the surrounding cells, resulting in aberrant expression of target genes such as CDKN1A, MMP9, and TGFB1. Previous study has revealed that CCL3/CCL4-CCR1 ligand-receptor pair has an effect on recruitment of MDSC, MSC, TAM and TAN into tumor niche and promote the expression of VEGF, resulting in angiogenesis9. Moreover, some compelling studies have reported that up-regulated genes induced by TGFB1 activity in stromal cell are reliable biomarkers of CRC metastasis and recurrence, and that TGFB1 signaling in cancer-associated fibroblasts (CAFs) enhances diffuse-type gastric cancer to invade extracellular matrix (ECM) and lymphatic vessels49,50. MMP9 is a secretory endopeptidase which has a non-negligible impact on ECM remodeling, EMT, new blood vessel formation, as well as immune response51. It inspired us that inhibition of these ligand-receptor bindings may be clinical targets to suppress tumor metastasis, impede the recruitment of immunosuppressive cells and potentiate response to ICIs.
To improve our understanding of CRC heterogeneity, we also predicted CMS of CRC samples. As a powerful classification system of CRC, CMS can act as the reference for feature clinical stratification and subgroup-based individualized therapy17. Based on previous report, there were 4 CMSs with distinguishing clinical features. The specific features are as follows: CMS1 (MSI immune) is characterized with MSI, hypermutation, immune infiltration and activation; CMS2 (Canonical) is characterized with WNT and MYC activation; CMS3 (Metabolic) is characterized with metabolic deregulation; CMS4 (Mesenchymal) is characterized with stromal infiltration, angiogenesis and TGF-β activation. Compared to the low-risk group, the high-risk group had a greater proportion of CMS4 in CRC samples. Furthermore, we found the expression levels of immune biomarker genes in CRC samples from the low-risk group were higher than those from the high-risk group. And interferon-α (IFN-α) response, interferon-γ (IFN-γ) response were enriched in CRC samples from the low-risk group. Homann et al. have reported that IFN-γ secreted by stimulated CD4+ T helper 1 cells can induce senescence in tumor cells in melanoma52. CRC samples in the high-risk group were characterized by hypoxia and EMT. Hypoxia is a hallmark of locally advanced tumors, in which lack of vascularization deprives cells of oxygen. In addition, hypoxia can also promote tumor progression and is one of the markers of poor prognosis53,54. The study of Otero-Albiol et al. has reported that hypoxia can facilitate bypass of senescence by supporting malignant transformation and inducing stemness properties, which enhances development and aggressiveness of cancer55. In terms of EMT, it has been reported that the onset of EMT is often accompanied by the bypass of senescence. And EMT also contributes to drug resistance of malignant cells56. Thus, we further explored the association between drug treatment and SRS of CRC samples. We found CRC samples with resistance to chemotherapy had higher SRS. At the same time, the predicted IC50 of 5-FU was significantly lower in CRC samples from the low-risk group than those from the high-risk group, suggesting that the low-risk samples had greater sensitivity to 5-FU based chemotherapy. This phenomenon may be caused by therapy-induced senescence (TIS), which could happen in both normal and tumor cells after treatment with anti-cancer chemotherapy agents57,58,59. In colon cancer cell line (HCT-116), Tato-Costa et al. found that 5-FU could promote senescence through enhancing expression of Slug and simultaneous stimulating EMT signal in a paracrine manner60. In addition to chemotherapy, we also found that there were significant differences of responses to immunotherapy between low- and high-risk groups. Anti-cancer immune response requires inter-collaboration between different immune cell populations. However, immunosenescence diminishes the immune response to cancer because of decreased naïve immune cells and increased memory immune cells61,62. Hence, SRS perhaps contribute to better classify CRC samples and provide a basis for individualized treatment.
Conclusions
Taken together, there are distinguishing biological characteristics between different senescence patterns of CRC. Based on these senescence patterns, we developed a senescence-related model for comprehensively exploring the heterogeneity of each CRC sample. After a series of systematic analysis of bulk and scRNA-seq, this model provides novel perspectives for exploring the mechanisms of tumor progression, TME remodeling, and response to ICIs in CRC.
Data availability
The public datasets analysed during the current study are available in the [TCGA] [https://portal.gdc.cancer.gov/] and [GEO] [https://www.ncbi.nlm.nih.gov/geo/]. Further inquiries are available from the corresponding author on reasonable request.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Siegel, R. L. et al. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 70, 145–164. https://doi.org/10.3322/caac.21601 (2020).
Gorgoulis, V. et al. Cellular senescence: Defining a path forward. Cell 179, 813–827. https://doi.org/10.1016/j.cell.2019.10.005 (2019).
Rhinn, M., Ritschka, B. & Keyes, W. M. Cellular senescence in development, regeneration and disease. Development https://doi.org/10.1242/dev.151837 (2019).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733. https://doi.org/10.1016/j.devcel.2014.11.012 (2014).
Pérez-Mancera, P. A., Young, A. R. J. & Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 14, 547–558. https://doi.org/10.1038/nrc3773 (2014).
Chandra, T. et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 10, 471–483. https://doi.org/10.1016/j.celrep.2014.12.055 (2015).
Guerrero, A. et al. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 19, e13133. https://doi.org/10.1111/acel.13133 (2020).
Korbecki, J. et al. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 21, 8412. https://doi.org/10.3390/ijms21218412 (2020).
Yang, X. et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer 19, 46. https://doi.org/10.1186/s12943-020-1146-4 (2020).
Zhang, E. et al. Exploring the relationship between lactate metabolism and immunological function in colorectal cancer through genes identification and analysis. Front. Cell Dev. Biol. 11, 1173803. https://doi.org/10.3389/fcell.2023.1173803 (2023).
Jolliffe, I. T. & Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 374, 20150202. https://doi.org/10.1098/rsta.2015.0202 (2016).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558. https://doi.org/10.1038/s41591-018-0136-1 (2018).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Van Calster, B. et al. Reporting and interpreting decision curve analysis: A guide for investigators. Eur. Urol. 74, 796–804. https://doi.org/10.1016/j.eururo.2018.08.038 (2018).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356. https://doi.org/10.1038/nm.3967 (2015).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. https://doi.org/10.1038/nature25501 (2018).
Rosenberg, J. E. et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920. https://doi.org/10.1016/S0140-6736(16)00561-4 (2016).
Şenbabaoğlu, Y. et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 17, 231 (2016).
Xu, L. et al. TIP: A web server for resolving tumor immunophenotype profiling. Cancer Res. 78, 6575–6580. https://doi.org/10.1158/0008-5472.CAN-18-0689 (2018).
Chen, D. S. & Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 39, 1–10. https://doi.org/10.1016/j.immuni.2013.07.012 (2013).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Natu. Commun. 4, 2612. https://doi.org/10.1038/ncomms3612 (2013).
Zhang, X. et al. Cell Marker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728. https://doi.org/10.1093/nar/gky900 (2019).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162. https://doi.org/10.1038/s41592-019-0667-5 (2020).
Mini, E. et al. RNA sequencing reveals PNN and KCNQ1OT1 as predictive biomarkers of clinical outcome in stage III colorectal cancer patients treated with adjuvant chemotherapy. Int. J. Cancer 145, 2580–2593. https://doi.org/10.1002/ijc.32326 (2019).
Lu, X. et al. Immune signature-based subtypes of cervical squamous cell carcinoma tightly associated with human papillomavirus type 16 expression, molecular features, and clinical outcome. Neoplasia 21, 591–601. https://doi.org/10.1016/j.neo.2019.04.003 (2019).
Ulloa-Montoya, F. et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J. Clin. Oncol. 31, 2388–2395. https://doi.org/10.1200/JCO.2012.44.3762 (2013).
Yang, C. et al. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: An in silico strategy towards precision oncology. Brief Bioinform. https://doi.org/10.1093/bib/bbaa164 (2021).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. https://doi.org/10.1038/s41586-019-1186-3 (2019).
Courtois-Cox, S., Jones, S. L. & Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 27, 2801–2809. https://doi.org/10.1038/sj.onc.1210950 (2008).
Price, J. S. et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell 1, 57–65 (2002).
Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 120, 513–522 (2005).
D’adda di Fagagna, F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522. https://doi.org/10.1038/nrc2440 (2008).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Blasco, M. A. et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34 (1997).
Counter, C. M. et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 11, 1921–1929 (1992).
Calcinotto, A. et al. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 99, 1047–1078. https://doi.org/10.1152/physrev.00020.2018 (2019).
Davaapil, H., Brockes, J. P. & Yun, M. H. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 144, 106–114. https://doi.org/10.1242/dev.138222 (2017).
Di Mitri, D. & Alimonti, A. Non-cell-autonomous regulation of cellular senescence in cancer. Trends Cell Biol. 26, 215–226. https://doi.org/10.1016/j.tcb.2015.10.005 (2016).
Toso, A., Di Mitri, D. & Alimonti, A. Enhancing chemotherapy efficacy by reprogramming the senescence-associated secretory phenotype of prostate tumors: A way to reactivate the antitumor immunity. Oncoimmunology 4, e994380 (2015).
Eggert, T. et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30, 533–547. https://doi.org/10.1016/j.ccell.2016.09.003 (2016).
Di Mitri, D. et al. Reversible senescence in human CD4+CD45RA+CD27− memory T cells. J. Immunol. 187, 2093–2100. https://doi.org/10.4049/jimmunol.1100978 (2011).
Lanna, A. et al. IFN-α inhibits telomerase in human CD8+ T cells by both hTERT downregulation and induction of p38 MAPK signaling. J. Immunol. 191, 3744–3752. https://doi.org/10.4049/jimmunol.1301409 (2013).
Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268. https://doi.org/10.1038/nri3175 (2012).
Adrover, J. M. et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity 50, 390–402. https://doi.org/10.1016/j.immuni.2019.01.002 (2019).
Bao, Z. et al. SNAIL induces EMT and lung metastasis of tumours secreting CXCL2 to promote the invasion of M2-type immunosuppressed macrophages in colorectal cancer. Int. J. Biol. Sci. 18, 2867–2881. https://doi.org/10.7150/ijbs.66854 (2022).
Wang, H. et al. KIAA1199 drives immune suppression to promote colorectal cancer liver metastasis by modulating neutrophil infiltration. Hepatology 76, 967–981. https://doi.org/10.1002/hep.32383 (2022).
Calon, A. et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 47, 320–329. https://doi.org/10.1038/ng.3225 (2015).
Ishimoto, T. et al. Activation of transforming growth factor beta 1 signaling in gastric cancer-associated fibroblasts increases their motility, via expression of rhomboid 5 homolog 2, and ability to induce invasiveness of gastric cancer cells. Gastroenterology 153, 191–204. https://doi.org/10.1053/j.gastro.2017.03.046 (2017).
Augoff, K., Hryniewicz-Jankowska, A., Tabola, R. & Stach, K. MMP9: A tough target for targeted therapy for cancer. Cancers 14, 1847. https://doi.org/10.3390/cancers14071847 (2022).
Homann, L. et al. IFN-γ and TNF induce senescence and a distinct senescence-associated secretory phenotype in melanoma. Cells 11, 1514. https://doi.org/10.3390/cells11091514 (2022).
Qiu, G.-Z. et al. Reprogramming of the tumor in the hypoxic niche: The emerging concept and associated therapeutic strategies. Trends Pharmacol. Sci. 38, 669–686. https://doi.org/10.1016/j.tips.2017.05.002 (2017).
Carnero, A. & Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. BioEssays 38(Suppl 1), S65–S74. https://doi.org/10.1002/bies.201670911 (2016).
Otero-Albiol, D. & Carnero, A. Cellular senescence or stemness: hypoxia flips the coin. J. Exp. Clin. Cancer Res. CR 40, 243. https://doi.org/10.1186/s13046-021-02035-0 (2021).
Faheem, M. M. et al. Convergence of therapy-induced senescence (TIS) and EMT in multistep carcinogenesis: Current opinions and emerging perspectives. Cell Death Discov. 6, 51. https://doi.org/10.1038/s41420-020-0286-z (2020).
DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. https://doi.org/10.1038/nature10189 (2011).
Ewald, J. A., Desotelle, J. A., Wilding, G. & Jarrard, D. F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 102, 1536–1546. https://doi.org/10.1093/jnci/djq364 (2010).
Soto-Gamez, A. & Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 22, 786–795. https://doi.org/10.1016/j.drudis.2017.01.004 (2017).
Tato-Costa, J. et al. Therapy-induced cellular senescence induces epithelial-to-mesenchymal transition and increases invasiveness in rectal cancer. Clin. Colorectal Cancer 15, 170–178. https://doi.org/10.1016/j.clcc.2015.09.003 (2016).
Thomas, R., Wang, W. & Su, D.-M. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun. Ageing 17, 2. https://doi.org/10.1186/s12979-020-0173-8 (2020).
Aiello, A. et al. Immunosenescence and its hallmarks: How to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 10, 2247. https://doi.org/10.3389/fimmu.2019.02247 (2019).
Funding
This work was supported by the National Natural Science Foundation of China (No.81802933 to Xiao Yang); Science and Technology Commission of Shanghai Municipality (22ZR1439700 to Xiao Yang); and Guangci Distinguished Young Scholars Training Program (GCQN-2019-A07 to Xiao Yang).
Author information
Authors and Affiliations
Contributions
C.D., X.X. and X.Z. designed the study and collected research data, E.Z., S.L., X.F. were responsible for the interpretation of the results, C.D., X.X. analyzed data and drafted the manuscript, X.Z. revised the manuscript, J.M., X.Y. and L.Z. provided the overall guidance. All authors read and approved the final Manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, C., Xu, X., Zhang, X. et al. Investigating the role of senescence biomarkers in colorectal cancer heterogeneity by bulk and single-cell RNA sequencing. Sci Rep 14, 20083 (2024). https://doi.org/10.1038/s41598-024-70300-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70300-1
