
Abstract
Domain identification is a critical problem in spatially resolved transcriptomics data analysis, which aims to identify distinct spatial domains within a tissue that maintain both spatial continuity and expression consistency. The degree of coupling between expression data and spatial information in different datasets often varies significantly. Some regions have intact and clear boundaries, while others exhibit blurred boundaries with high intra-___domain expression similarity. However, most ___domain identification methods do not adequately integrate expression and spatial information to flexibly identify different types of domains. To address these issues, we introduce Spot2vector, a computational framework that leverages a graph-enhanced autoencoder integrating zero-inflated negative binomial distribution modeling, combining both graph convolutional networks and graph attention networks to extract the latent embeddings of spots. Spot2vector encodes and integrates spatial and expression information, enabling effective identification of domains with diverse spatial patterns across spatially resolved transcriptomics data generated by different platforms. The decoders enable us to decipher the distribution and generation mechanisms of data while improving expression quality through denoising. Extensive validation and analyses demonstrate that Spot2vector excels in enhancing ___domain identification accuracy, effectively reducing data dimensionality, improving expression recovery and denoising, and precisely capturing spatial gene expression patterns.
Similar content being viewed by others


Introduction
Understanding the spatial distribution of cells within tissues is crucial for elucidating their biological functions and characterizing their interaction patterns, as the relative positions of cells significantly impact cell behavior and tissue characteristics1,2,3. Recent advancements in spatially resolved transcriptomics (ST), such as 10X Visium4, Stereo-seq5, Slide-seq6, and MERFISH7, have revolutionized our ability to measure gene expression within the spatial context of tissue architecture8,9. These cutting-edge technologies overcome the limitations of single-cell transcriptomics (that is, single-cell RNA sequencing, scRNA-seq)10 by providing extra spatial information, offering unprecedented insights into tissue spatial heterogeneity and cellular communication11,12. They help reveal complex tissue structures and functions13,14, enable the tracking of cell fate and development5,15, and significantly advance our understanding of complex biological systems16,17.
Identifying spatial domains is one of the most important topics in ST research. It involves deciphering distinct spatially continuous regions in tissues with similar expression patterns3. Existing ___domain identification methods can be broadly classified into two categories based on the utilization of spatial information: non-spatial and spatial clustering methods. Traditional clustering methods developed for scRNA-seq data, such as K-means18, and those implemented in the Scanpy toolkit based on Louvain or Leiden algorithms19,20,21, can also be applied to ST data. However, these methods rely only on gene expression data and do not incorporate spatial information, thus the identified domains will lack spatial continuity. In contrast, spatial clustering methods specifically designed for ST data aim to effectively integrate spatial information with gene expression data, providing a deeper understanding of the spatial architecture and its biological implications. For example, SpaGCN designs a graph convolutional network to integrates gene expression, spatial ___location and histology information, and uses an unsupervised iterative clustering algorithm to identify spatial domains2. STAGATE employs a graph attention auto-encoder network to learns low-dimensional latent embeddings of spots by adaptively aggregating information from its spatial neighbors3. Similarly, DeepST utilizes a combination of a graph neural network autoencoder and a denoising autoencoder to jointly produce a latent representation of augmented ST data1. GraphST integrates graph neural networks with augmentation-based self-supervised contrastive learning strategy to obtain spot embeddings22. Besides, stLearn uses a spatial graph-based neural network to correct for technical noise and performs unsupervised clustering for ST data23.
While current approaches have proven useful in identifying spatial domains, they still lack efficiency in addressing several critical challenges. A key limitation lies in the inflexibility of processing and integrating expression and spatial information, whose relative importance in the ___domain identification task varies across different ST datasets. For instance, in tissues such as the human brain24, regions exhibit clear boundaries and distinct layers, making spatial continuity more important for ___domain identification. In contrast, in tissues such as tumor-invaded breast25, ___domain boundaries are less delineated, making the similarity of expression information more crucial. This variability necessitates a more flexible strategy for handling different ___domain annotations and data platforms. Additionally, the inherent noise in ST data is compounded not only by limited sequencing depth but also by the delicate experimental steps required to preserve spatial locations16,26, further complicating the extraction of meaningful signals from technical artifacts. It is essential not only to denoise the data by restoring expected expression levels and estimating dropout rates, but also to accurately reconstruct the spatial expression patterns of genes. Overcoming these obstacles is vital for revealing the underlying biological significance within ST data, and helping to interpret the intricate underlying biological mechanisms within tissues.
To address the aforementioned challenges, we proposed a computational method, Spot2vector, to extract low-dimensional embeddings of spots using a zero-inflated negative binomial (ZINB)-based graph-enhanced autoencoder model. Spot2vector integrates both Graph Convolutional Networks (GCN) and Graph Attention Networks (GAT) within its encoder framework, which enables efficient aggregation of neighborhood information and adaptive adjustment of graph edge weights, as well as ensures computational efficiency. Spot2vector encodes spatial and expression information separately, and adjusts their relative importance through a tunable parameter \(\lambda\) to accommodate different types of domains. This approach allows for a flexible preservation of both types of crucial information across various datasets and ___domain annotations, thereby extracting effective low-dimensional representations of spots according to users’ prior knowledge. The decoder framework of Spot2vector is designed to output the parameters for the ZINB distribution, and characterize the ST data by optimizing the maximum likelihood objective. This strategy not only deepens our understanding of the generation mechanisms underlying ST data, but also contributes to denoising the expression data to improve its quality.
We have demonstrated through sufficient experiments that Spot2vector facilitates superior analysis of ST data generated by different platforms. This includes improving ___domain identification performance to better elucidate spatial heterogeneity, integrating spatial and expression information more flexibly to better accommodate diverse ___domain labels and datasets, denoising gene expression to better portray their spatial patterns, and identifying subdomains to better reveal finer biological details. Our studies suggest that Spot2vector is a promising tool to comprehensively analyze the ST data.
Results
Overview of Spot2vector
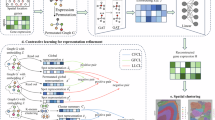
Spot2vector leverages a ZINB-based graph-enhanced autoencoder model to extract low-dimensional representations of ST data, effectively integrating spatial and expression information (Fig. 1). Specifically, Spot2vector first performs basic data preprocessing steps, including normalization and the screening of highly variable genes (HVGs) (Methods). It then constructs a spatial graph \({G}_{S}\) and an expression graph \({G}_{E}\), based on the relative spatial coordinates \(C\,{{\mathbb{\in }}\,{\mathbb{R}}}^{S* 2}\) of spots and the gene expression matrix \(X\in {{\mathbb{R}}}^{S* G}\), respectively (Fig. 1a, Methods), where \(S\) is the number of spots and \(G\) is the number of genes. The network architecture of Spot2vector utilizes GCN and GAT as encoders, with three-layer multilayer perceptron (MLP) as decoders (Fig. 1b). Spot2vector includes two independent graph encoders, one combining the expression matrix with spatial proximity information, and the other with gene expression similarity for encoding. Through these two graph encoders, Spot2vector generates two complementary spot embeddings, \({Z}_{S}\) and \({Z}_{E}\), which are then linearly combined using an adjustable parameter \(\lambda\) (referred to as \({\lambda }_{{{{\rm{train}}}}}\) during training) to produce the integrated spot low-dimensional representations \(Z\). Three MLP decoders generate three parameter matrices- \({{{\rm{ M}}}}\), \({{\Theta }}\), and \({{\Pi }}\) -from the low-dimensional representation of spots, corresponding to the expectation, dispersion, and zero-inflated probability of the ZINB distribution, respectively (Methods). This strategy enables us to interpret the distribution and generation mechanisms of ST data, and denoise the expression data to enhance its quality. Notably, the linear combination parameter \(\lambda\) can be further adjusted during the inference process (referred to as \({\lambda }_{{{{\rm{infer}}}}}\)). Setting \({\lambda }_{{{{\rm{infer}}}}}\) to 0 means the model will primarily focus on spatial proximity information, while setting it to 1 emphasizes expression similarity information. (Fig. 1c). This flexibility allows for tailored integration of the two types of information in the final low-dimensional representation of spots. The output of Spot2vector can be utilized for various downstream analyses, including ___domain identification tasks that prioritize spatial continuity, cell type clustering that emphasizes expression consistency, gene expression recovery, and subdomain division (Fig. 1d).

a Spot2vector first constructs a spatial graph and an expression graph based on the relative spatial ___location of spots and the gene expression matrix, respectively. b Spot2vector employs ZINB-based graph-enhanced autoencoder to extract low-dimensional representations of spots, using GCN and GAT as encoders and three-layer MLP as decoders. c The linear combination parameter can be further adjusted during the inference process: setting it to 0 focuses on spatial information, while setting it to 1 focuses on expression information. d The output of Spot2vector can be used for various downstream analyses.
Spot2vector demonstrates superior ___domain identification performance
To assess the efficacy of Spot2vector in ___domain identification, we applied it to several ST datasets with annotated ___domain labels. These datasets were generated using various platforms with differing resolutions, including 10X Visium, Stereo-seq, and MERFISH, enabling a comprehensive evaluation of method performance (Supplementary Note 1, Supplementary Table S1). The spot embeddings derived from Spot2vector were employed for unsupervised spatial clustering using the mclust algorithm27 (Methods). We then assessed the alignment of the predicted ___domain labels with the annotated labels using adjusted rand index (ARI) and normalized mutual information (NMI) metrics. Spot2vector was benchmarked against six state-of-the-art ___domain identification methods: Scanpy19, Stlearn23, SpaGCN2, GraphST22, DeepST1 and STAGATE3 (Supplementary Note 2).
Across all tested ST datasets, Spot2vector consistently demonstrated superior ___domain identification performance (Fig. 2a, Supplementary Figs. S1, S2). The human dorsolateral prefrontal cortex (DLPFC) dataset24 is recognized as a standard benchmark for evaluating ___domain identification methods, consisting of 12 sections. The ___domain annotations were primarily derived based on histological organization and cytoarchitecture, emphasizing spatial continuity. Spot2vector achieved the highest ___domain identification accuracy on this dataset (Fig. 2b, Supplementary Figs. S3-S5). For instance, Spot2vector successfully identified the spatial hierarchical structure of human brain in section 151672, while other methods disrupted the spatial continuity of domains to varying degrees, particularly in “Layer_3” (Fig. 2c). Additionally, Spot2vector offers several tunable parameters, including the integration strength between expression and spatial information during the training phase (\({\lambda }_{{{{\rm{train}}}}}\)), and the average degree of nodes in the spatial and expression graphs (\({k}_{{GS}}\) and \({k}_{{GE}}\)). Our results demonstrate that Spot2vector consistently outperforms STAGATE in ___domain identification performance across a wide range of parameter combinations, even when the tunable parameters of STAGATE are varied (Supplementary Fig. S6).

a Performance comparison of seven ___domain identification methods (Spot2vector, DeepST, SpaGCN, Scanpy, Stlearn, STAGATE, GraphST) tested on six ST datasets, using ARI as the evaluation metric. b ARI scores (x-axis) of seven methods (y-axis) tested on 12 DLPFC sections. Each box plot ranges from the first and third quartiles with the median as the vertical line, while whiskers represent 1.5 times the interquartile range from the lower and upper bounds of the box. Data beyond the end of the whiskers are plotted individually. c Spatial plots of ___domain annotation, and seven methods on the section 151676 of DLPFC dataset. Each method colors the spot with their predictive ___domain labels. d Spatial plots of ___domain annotation, and seven methods on the Mouse Brain dataset. Three regional boundaries are framed by dashed lines: yellow (Fiber_tract), black (Hypothalamus_2), white (Hypothalamus_1).
Additionally, Spot2vector outperformed all other methods (ARI = 0.70) in ___domain identification on the Mouse Brain 10X Visium dataset (Fig. 2d). Among the 15 predicted categories, Spot2vector accurately identified a majority of brain structures with various shapes, including hooked areas (e.g., Fiber_tract), flat areas (e.g., Hypothalamus_2), and block-like areas (e.g., Hypothalamus_1). However, other methods failed to effectively identify these three areas. STAGATE, which primarily relies on spatial information, mostly generates block-like structures. In contrast, Scanpy, which overly depends on expression information, produces blurred boundaries and lacks clear spatial patterns.
To sum up, Spot2vector demonstrates superior performance in ___domain identification across all benchmark tests and is capable of preserving fine-grained spatial hierarchical structures, proving its robustness and versatility for better ST data analysis.
Spot2vector adapts to different annotations for accurate ___domain and cell type clustering
There may be multiple spot annotations for the same ST dataset, with some emphasizing spatial continuity within domains and others focusing on expression similarity within classes. Consequently, spatial clustering tasks require careful handling to flexibly adapt to different annotations.
The Allen Mouse Brain Aging (AMBA) MERFISH dataset generated a cell atlas of mouse brain at single-cell resolution28. This high-resolution ST dataset includes both cell type annotations (Fig. 3a) and spatial ___domain annotations (Fig. 3b). We applied seven methods, including Spot2vector, to this dataset and assessed their predictive performance across different annotations. Our analysis reveals that when the ground truth labels are based on intra-class expression similarity, methods, such as SpaGCN, Stlearn, DeepST and Scanpy demonstrate superior performance, whereas GraphST and STAGATE are less effective (Fig. 3a, Supplementary Fig. S7). Conversely, when the ground truth labels favor intra-class spatial continuity, the performance trends are reversed (Fig. 3b, Supplementary Fig. S8). This indicates that existing methods typically excel only under a specific labeling paradigm and lack the flexibility to accommodate alternative labeling schemes. In contrast, Spot2vector exhibits strong adaptability by adjusting the value of parameter \({\lambda }_{{{{\rm{infer}}}}}\) for different annotations (Supplementary Fig. S9). This allows it to derive low-dimensional embeddings that fully integrate spatial and expression information, thereby achieving outstanding clustering performance for both annotations (Fig. 3c, Supplementary Fig. S10).

a, b The spatial clustering performance of various methods, using the (a) cell type annotation and (b) ___domain annotation as ground truth labels, respectively. The spatial plots are colored by the predicted labels of various methods, while the UMAP plots are colored by the annotation labels. c The NMI scores (y-axis) of seven methods (x-axis) under cell type (CT) and ___domain annotations. d The correlation matrix of cell type distribution under true ___domain labels and Spot2vector predicted ___domain labels, as well as the correlation matrix of ___domain distribution under true cell type labels and Spot2vector predicted cell type labels. The color bar goes from blue (negative correlation) to red (positive correlation), corresponding to the magnitude of the correlation coefficient. e Same as d, showing the correlation matrices between true domains and between true cell types.
Additionally, we observed that one spatial ___domain typically contains various cell types, while the same cell types can be distributed across multiple spatial domains (Supplementary Fig. S11). Accurately deciphering the spatial distribution of cell types and the cellular composition of spatial domains is essential for understanding tissue organization. The results presented in Fig. 3d and Fig. 3e highlight Spot2vector’s exceptional performance in both tasks. The correlation between cell type distribution within the domains predicted by Spot2vector and the true domains strongly matches the correlation observed among the true domains themselves. Spot2vector also achieves remarkable accuracy in predicting the spatial distribution of cell types within these domains. In contrast, other methods, such as STAGATE, GraphST, and DeepST, fail to perform well across both tasks (Supplementary Figs. S12-S14). For instance, neuron cells predominantly localize within three cortical layer regions (V, VI, II/III), which exhibit similar cell type distributions (Supplementary Fig. S11). Spot2vector accurately predicts all three domains, with regions 6, 2, and 5 corresponding to cortical layers V, VI, and II/III, respectively. Spot2vector also precisely identifies neuron cells (cluster 4 in Fig. 3a, d and e) and their spatial ___domain distribution. In contrast, other methods either fail to distinguish neuron cells and their ___domain distributions (e.g., STAGATE, Supplementary Fig. S12), or struggle to identify cortical layers and their cell type distributions (e.g., DeepST, Supplementary Fig. S14). These results highlight Spot2vector’s superiority in accurately predicting cell type distribution and ___domain composition.
Spot2vector enables flexible integration of spatial and expression information
To further illustrate Spot2vector’s ability to flexibly integrate spatial and expression information, we applied Spot2vector to the Mouse Organogenesis Spatiotemporal Transcriptomic Atlas (MOSTA) dataset generated using Stereo-seq technology5 (Fig. 4a). This dataset comprises 12 domains corresponding to various mouse organs, annotated with cluster-specific marker genes and validated by ___domain expertise. Comparison of ___domain identification results shows that Spot2vector uniquely identified several critical regions, such as Cavity (cluster 2) and Neural crest (cluster 10), demonstrating superior identification performance (Fig. 4b, Supplementary Fig. S15). After model training is complete, the parameter \({\lambda }_{{{{\rm{infer}}}}}\) can be further adjusted to flexibly integrate expression and spatial information (Methods). As expected, when the parameter \({\lambda }_{{{{\rm{infer}}}}}\) was set to 0, spatial information predominated, resulting in more continuous predicted domains and exhibiting higher spatial consistency. Conversely, when \({\lambda }_{{{{\rm{infer}}}}}\) was set to 1, expression information became dominant, leading to predicted domains that prioritized intra-___domain expression similarity. As \({\lambda }_{{{{\rm{infer}}}}}\) was gradually adjusted from 0 to 1, the identified domains progressively transcend rigid boundaries, showcasing more flexible clustering outcomes. Notably, when \({\lambda }_{{{{\rm{infer}}}}}\) was set to 0.9, the model achieved an optimal balance in spatial clustering (Fig. 4c).

a Domain annotation of the MOSTA Stereo-seq dataset (E9.5). b Spatial clustering results from five methods (SpaGCN, DeepST, GraphST, STAGATE and Spot2vector), colored by the predicted labels of corresponding methods. c The left-to-right panels illustrate progressive changes in Spot2vector predictions as the parameter \({\lambda }_{{{{\rm{infer}}}}}\) is tuned from 0 (blue arrowheads) to 1 (red arrowheads). d Four mouse embryonic regions (Brain, Cavity, Heart, Neural crest), and the spatial expression patterns of eight corresponding SVGs (two per region) before and after Spot2vector denoising.
Moreover, Spot2vector infers the parameters of the ZINB distribution for expression data within its decoder network. Therefore, it can interpret the original expression data based on mathematical models, thereby achieving effective data denoising (Methods). Through differential expression (DE) analysis, we identified several spatially variable genes (SVGs) across four regions and compared their expression patterns before and after Spot2vector denoising. We found that these genes showed more significant spatial expression patterns after Spot2vector denoising, and corresponded precisely to their respective regions (Fig. 4d, Supplementary Fig. S16). For genes with dense raw expression, such as gene Hbb-y, Spot2vector corrected the original expression to produce biologically meaningful patterns. For genes with sparse raw expression, such as Actc1, Spot2vector effectively filled in missing dropout values while recovering count values, significantly enhancing the clarity of gene expression pattern. These results demonstrate that Spot2vector has highly effective denoising capabilities, enhancing the clarity and biological relevance of the identified SVGs within each ___domain.
Spot2vector identifies biologically meaningful spatial subdomains in breast cancer
To further evaluate the capabilities of Spot2vector in ___domain identification, we applied it to the human breast cancer 10X Visium dataset and conducted detailed analyses of the intricate subdomains and biomarkers discovered. This dataset was manually annotated into four categories based on H&E-stained images: Ductal Carcinoma In Situ/Lobular Carcinoma In Situ (DCIS/LCIS), Invasive Ductal Carcinoma (IDC), healthy tissue, and tumor edge, resulting in a total of 20 distinct domains29 (Fig. 5a).

a Domain annotation of the human breast cancer 10X Visium dataset. b Spatial clustering results of five methods. c From top to bottom: reference histological images; domains 3 and 10 predicted by Spot2vector superimposed on histology; high-contrast enhancement of histological features within the predicted domains. d,e: (d) The raw and (e) Spot2vector-denoised gene expression differences of marker genes between subdomains 3 (n = 110) and 10 (n = 349). A two-sided Wilcoxon Rank Sum test is used to test the difference. f Tumor and tumor edge annotations of the dataset. g,h: (g) The raw and (h) Spot2vector-denoised gene expression differences of marker genes between the tumor (n = 2490) and tumor edge (n = 823). A two-sided Wilcoxon Rank Sum test is used to test the difference. All box plots range from the first and third quartiles with the median as the horizontal line, while whiskers represent 1.5 times the interquartile range from the lower and upper bounds of the box. i Spatial plots of marker genes, using their raw expression (upper panels) and Spot2vector-denoised expression (lower panels), respectively. j Survival analyses of four genes. A two-sided Log-rank test is used to compare the differences in survival curves between groups. HR stands for Hazard Ratio.
Consistent with previous results, Spot2vector outperformed all evaluated ___domain identification methods in terms of identification accuracy (Fig. 5b, Supplementary Fig. S17). Further analysis of Spot2vector’s ___domain identifications revealed that the ___domain originally labeled as “IDC_2” was segmented into subdomains 3 and 10 (Fig. 5c). To elucidate the biological significance of these subdomains, we conducted DE analysis, which successfully identified several marker genes for each subdomain, highlighting their distinct biological characteristics (Fig. 5d). Specifically, the Invasive Core Subtype (subdomain 10) was marked by genes associated with high invasiveness and proliferation, suggesting a more aggressive phenotype with enhanced metastatic potential. For example, MUC19 contributes to cancer cell survival and drug resistance when overexpressed, thereby promoting tumor progression30,31. LTO1 is associated with breast cancer by influencing the immune microenvironment. Higher expression of LTO1 correlates with more aggressive tumor behavior and poorer clinical outcomes32. Similarly, ABCC11, a transporter linked to drug resistance in various cancers, particularly breast cancer33,34,35, was found to be elevated in more aggressive subtypes, correlating with lower disease-free survival rates36,37 (Supplementary Fig. S18). In contrast, the Immune-Frontier Subtype (subdomain 3) was characterized by immune-related genes, indicating an active immune microenvironment at the tumor boundary. For instance, IGLC3 is closely associated with antibody production38, high expression of IGLC3 at tumor boundaries may be implicated in immune evasion mechanisms, where tumor cells alter the activity of surrounding immune cells to evade detection and destruction39,40(Supplementary Fig. S18). CCDC80, another key marker, regulates cell migration and adhesion41, and is strongly associated with immune cell infiltration in the tumor microenvironment42,43. SFRP4 is known for its involvement in the Wnt signaling pathway, reduced expression of SFRP4 can lead to increased Wnt signaling activity, promoting tumor growth and metastasis44,45. Overall, these marker genes revealed the subdomains’ distinct biological roles in tumor microenvironment, further highlighting Spot2vector’s effectiveness in identifying and differentiating complex domains.
We also compared the differential expression of these marker genes in the raw and Spot2vector-denoised expression data between two subdomains, and demonstrated more significant differences in the denoised data (Fig. 5e, Supplementary Fig. S18). This not only demonstrate Spot2vector’s ability to reconstruct spatial gene expression patterns, but also underscore the potential biological significance of these marker genes. Furthermore, given that the ST data is originally annotated with both tumors (DCIS/LCIS and IDC) and discontinuous tumor edges (Fig. 5f), we further verified the differential expression of identified marker genes between tumor and tumor edge regions. As illustrated in Fig. 5g and Fig. 5h, SFRP4 and CCDC80 are highly expressed at the tumor edge, while MUC19 and LTO1 are highly expressed at the tumor region. It is noteworthy that Spot2vector denoising enhances the spatial expression patterns of marker genes, and clearly delineate the structures of the tumor and its boundary (Fig. 5i). Further survival analysis also confirmed that high expression of tumor region marker genes is associated with improved patient outcomes, while high levels of tumor marker genes are linked to poorer prognosis (Fig. 5j). These results highlight the reasonableness of the biological interpretation of Spot2vector’s subdomains, and demonstrates its capability in identifying fine structure of spatial domains.
Spot2vector enhances spatial gene expression patterns through effective denoising
The incorporation of the ZINB module in Spot2vector has proven effective in recovering biologically meaningful gene expression patterns. To further evaluate its denoising performance, we conducted a systematic comparison using the human breast cancer dataset, evaluating it against three widely used denoising methods \(-\) STAGATE3, scVI46, and MAGIC47. First, we performed DE analysis between tumor and healthy regions (Fig. 5f) based on the raw expression data, and identified the top 200 DE genes from each region, yielding a total of 400 marker genes. We then quantified the changes in -log(p-value) for these genes before and after applying each of the four denoising methods. While all methods enhanced the statistical significance of marker genes to some extent (Supplementary Fig. S19), Spot2vector achieved superior denoising performance compared to STAGATE (232 vs. 168), scVI (340 vs. 60), and MAGIC (288 vs. 112), as demonstrated in Fig. 6a.

a Scatterplots comparing the significance (-log(p-value)) of the marker genes after denoising using Spot2vector (x-axis) against raw expression, STAGATE, scVI, and MAGIC (y-axis). Each dot represents a gene (n = 400). b The top panels show the raw and denoised expression distributions of the MUC19 gene using four methods. The middle panels display the corresponding high-expression regions extracted using gene-specific thresholds, and the bottom panels show the gene expression values. c Ripley’s K and L curves of MUC19 gene across raw data and data denoised by four methods. d Boxplots compare the maximum Ripley’s K and L scores for 400 marker genes across raw data and denoised datasets. All box plots range from the first and third quartiles with the median as the horizontal line, while whiskers represent 1.5 times the interquartile range from the lower and upper bounds of the box. Data beyond the end of whiskers are plotted individually.
To further quantitatively evaluate the enhancement in gene expression pattern clarity after denoising, we first need to extract the gene expression patterns after denoising. We developed a heuristic approach that established gene-specific thresholds based on the distribution of expression values to extract spatial expression patterns (Methods, Fig. 6b). For example, while the MUC19 gene displayed no obvious spatial pattern in the raw expression data, all four denoising methods revealed more distinct spatial expression patterns characterized by continuous high-expression regions aligned with tumor areas and low-expression regions corresponding to healthy areas. This suggests that denoising effectively restored biologically relevant spatial expression features.
To quantify and compare the gene expression pattern enhancement effects after denoising using different methods, we calculated the Ripley’s K and L curves for each gene (Methods). Taking MUC19 as an example, Spot2vector consistently achieved the highest Ripley’s K and L scores across multiple radius (Fig. 6c), reflecting a robust high-expression pattern within tumor regions after denoising, which aligns with previously reported findings in the literature 30,31. Additionally, we applied Ripley’s K and L functions to all 400 marker genes and derived the maximum values from each curve as representative Ripley’s K and L scores. As shown in Fig. 6d, scVI is comparable to the original expression, while MAGIC, STAGATE, and Spot2vector scores are relatively higher, with Spot2vector being the highest. This indicates that Spot2vector is able to recover the clearest spatial gene expression pattern.
In summary, Spot2vector effectively removes noise and restores spatial gene expression patterns, outperforming other methods in enhancing marker gene significance and revealing biologically meaningful spatial features.
Discussion
The emergence of spatially resolved transcriptomics technology has created opportunities for exploring the spatial structure of tissues. However, the inherent complexity and heterogeneity of ST datasets present significant analytical challenges. The spatial continuity and expression similarity vary significantly across different domains in ST datasets, necessitating ___domain identification approaches capable of adapting and adjusting to these variations. In light of this, we introduce Spot2vector, a computational method that addresses these challenges by integrating spatial and expression information. Spot2vector employs a combination of the GCN and GAT models to encode ST data. It uses an MLP to decode the parameters of the ZINB distribution, providing a mathematical interpretation of observed gene expression.
One of the key strengths of Spot2vector lies in its ability to balance spatial continuity and expression similarity, which are often conflicting objectives in spatial clustering. By incorporating tunable parameters, Spot2vector allows researchers to tailor the analysis to specific dataset characteristics and research questions, thereby achieving optimal ___domain identification. This flexibility is crucial for accurately interpreting complex tissues, such as the human brain and cancerous tissues, where spatial domains often exhibit intricate patterns of heterogeneity.
The denoising capability of Spot2vector further enhances its utility in ST data analysis. By reconstructing gene expression data while mitigating the effects of dropout, Spot2vector produces more biologically meaningful representations of spatial gene expression patterns. This not only improves clustering performance but also facilitates the identification of spatially variable genes, providing deeper insights into the underlying biological processes.
Spot2vector employs L1 regularization in its objective function, applied to the dispersion parameters of the ZINB distribution. This promotes sparsity, encouraging some parameters to shrink to zero, which is particularly beneficial for identifying genes with significant variability across spatial domains. Comparative analyses demonstrate that L1 regularization consistently achieves superior ___domain identification accuracy relative to both L2,1 and L2 regularization terms (Supplementary Fig. S20). Furthermore, Spot2vector strategically integrates the complementary strengths of GCN and GAT architectures. While GCN effectively captures local graph structures through its fixed aggregation mechanism, it may be limited in modeling complex and non-linear relationships. In contrast, GAT employs attention mechanisms to dynamically weight neighboring nodes, enabling the capture of nuanced relationships at increased computational cost. By combining GCN and GAT, Spot2vector achieves a balance: GCN provides a robust foundation for learning local structures, while GAT enhances the model’s ability to capture complex relationships. Experiments on six benchmark ST datasets show that the GCN + GAT combination outperforms configurations using only GCN or GAT in ___domain identification accuracy. Additionally, the GCN + GAT framework maintains computational efficiency, with running times and memory usage scaling linearly with the number of spots and genes (Supplementary Figs. 21, 22).
Despite Spot2vector’s superiority in ___domain identification tasks compared to existing methods, there remains room for improvement. Currently, the selection of the hyperparameter \({\lambda }_{{{{\rm{infer}}}}}\) depends on the user’s prior knowledge and specific demands. Future work could focus on developing methods to automatically determine this parameter, or even explore dynamic weighting mechanisms, such as attention-based or trainable weight matrices, to adapt to dataset variability. Additionally, incorporating multi-omics information or including multi-level structures could enable more effective and comprehensive ___domain identification.
Conclusion
Spot2vector is a powerful tool for advancing ST research, offering superior performance in ___domain identification and data denoising. Its innovative integration of GCN, GAT, and ZINB models enables a flexible and robust analysis of ST data, accommodating diverse biological contexts and research objectives. By denoising expression data and revealing spatial patterns, Spot2vector facilitates a deeper understanding of complex tissue architectures and the biological mechanisms underlying spatial heterogeneity. This framework holds significant promise for enhancing our understanding of tissue organization, and disease pathology, paving the way for future discoveries in spatial transcriptomics and its applications in biomedical research.
Methods
Data preprocessing
The Spot2vector method takes ST data \(\left(X,{C}\right)\) as input, where \(X\in {{\mathbb{R}}}^{S* G}\) represents the gene expression matrix, and \(C\in {{\mathbb{R}}}^{S* 2}\) represents the spots ___location, \(S\) is the number of spots and \(G\) is the number of genes. Several preprocessing steps are applied to the raw ST data to ensure data quality and emphasize biologically relevant features. Specifically, the dataset was filtered to retain only those genes with more than 100 total counts across all spots, ensuring that only sufficiently expressed genes were included. Normalization was performed by scaling the total counts to 10,000 per spot, followed by a logarithmic transformation to stabilize the variance across different expression levels. Subsequently, HVGs were identified using the Seurat v3 method48, selecting the top 8,000 most variable genes by default. This step reduces the dataset’s dimensionality and focuses on genes that are most likely to capture the underlying biological variance.
Spatial and expression graph construction
To construct the spatial proximity graph \({G}_{S}\) and the expression similarity graph \({G}_{E}\), we utilized the spatial coordinates \(C\in {{\mathbb{R}}}^{S* 2}\) and gene expression profiles \(X\in {{\mathbb{R}}}^{S* G}\), respectively. In both graphs, nodes correspond to individual spots, while edges represent either spatial proximity or expression similarity between spots. The spatial graph \({G}_{S}\) is constructed using the radius-based method, implemented by the “radius_neighbors_graph” function from the “scikit-learn” Python library. Specifically, two spots are connected if their Euclidean distance is within a predefined radius \(r\). This approach captures the local spatial relationships between spots, ensuring that only physically proximate spots are connected in \({G}_{S}\). In addition, the expression graph \({G}_{E}\) is constructed using the \(k\)-nearest neighbors’ method, implemented by the “kneighbors_graph” function from the “scikit-learn” Python library. Specifically, each spot is connected to its \(k\) nearest neighbors based on expression similarity. This approach captures the functional relationships between spots, ensuring that spots with similar gene expression profiles are connected in \({G}_{E}\). The selection of \(r\) and \(k\) involves a trade-off between computational efficiency and the preservation of local structure. Empirically, we set these parameters such that the average node degree in the resulting graphs does not exceed 8 3,23. This guideline ensures that the graphs are sufficiently informative while avoiding excessive computational overhead. The specific values of \(r\) and \(k\) used for each dataset in our study are provided in Supplementary Table S2. To validate the robustness of Spot2vector under different values of \(r\) and \(k\), we conducted extensive tests by varying \(r\) and \(k\) across a range of values (from 3 to 8). The results demonstrate that Spot2vector’s performance remains stable under different parameter settings (Supplementary Fig. S6).
ZINB-based graph-enhanced autoencoder model
The network architecture of Spot2vector consists of two main components: the graph encoders and the MLP decoders. The graph encoder in Spot2vector is designed as a combination of the graph convolutional network (GCN) and the graph attention transformer (GAT).
Graph encoder
The GCN layers leverages the local neighborhood information in the graph to update node features. This process involves aggregating features from neighboring nodes to learn more robust representations. The standard GCN layer is mathematically defined as follows49:
where \({Z}^{\left(l\right)}\in {{\mathbb{R}}}^{S* {D}_{l}}\) is the input node feature matrix at layer \(l\), \({D}_{l}\) is the dimension of the feature at layer \(l\), and \({Z}^{\left(0\right)}=X\). \(\widetilde{A}=A+I\) is the adjacency matrix \(A\) with added self-connections (identity matrix \(I\)), and \(\widetilde{D}\) is the diagonal degree matrix of \(\widetilde{A}\), where \({\widetilde{D}}_{{ii}}={\sum}_{j}{\widetilde{A}}_{{ij}}.\quad {W}^{\left(l\right)}\) is the trainable weight matrix at layer \(l\), and \(\sigma\) is the ELU (Exponential Linear Unit) activation function to ensure stable training and faster learning.
The node features and graph structures output by GCN are input into GAT to further enhances the graph representation by applying attention mechanisms that allow for the adaptive weighting of edges. This attention mechanism assigns different importance to each neighbor, enabling the model to focus on more relevant features and improving the expressiveness of the learned embeddings. The update rule for GAT layer is defined as follows50:
where \({Z}_{i}^{\left(l+1\right)}\) is the updated feature representation of node \(i\) at layer \(l+1,\) and \({{{\mathscr{N}}}}\left(i\right)\) represents the set of neighbors of node \(i\). The attention coefficient \({\alpha }_{{ij}}^{\left(l\right)}\) is computed as:
where \(a\) is the learnable attention vector, \(\parallel\) denotes the concatenation operation, \({W}^{\left(l\right)}\) is the trainable weight matrix at layer \(l\), and LeakyReLU (Leaky Rectified Linear Unit) is the activation function used to introduce non-linearity.
Dual embeddings
Two separate graph encoders, \({E}_{S}\) and \({E}_{E}\), have the same network architecture and both receive the gene expression matrix \(X\) as input (initial node features), but are trained on different graphs \({G}_{S}\) and \({G}_{E}\). The dual encoder approach in Spot2vector generates two complementary embeddings \({Z}_{S}\) and \({Z}_{E}\) by leveraging information from spatial or expression neighbors. The embeddings derived from two encoders are then linearly combined using a tunable parameter \(\lambda\) (default as 0.5 during training, referred to as \({\lambda }_{{{{\rm{train}}}}}\)):
Here, \(\lambda\) can be further adjusted during the inference process (referred to as \({\lambda }_{{{{\rm{infer}}}}}\)) with fixed embeddings \({Z}_{S}\) and \({Z}_{E}\). When \({\lambda }_{{{{\rm{infer}}}}}\) equals 0, it generates embeddings entirely based on spatial neighbor information; when \({\lambda }_{{{{\rm{infer}}}}}\) equals 1, it generates embeddings entirely based on expression neighbor information. This allows Spot2vecotr to adjust the low-dimensional embeddings according to different needs, ensuring the retention of critical information specific to various datasets.
Note that \({\lambda }_{{{{\rm{train}}}}}\) and \({\lambda }_{{{{\rm{infer}}}}}\) serve distinct roles in our framework. \({\lambda }_{{{{\rm{train}}}}}\) is fixed during training to ensure a balanced integration of expression and spatial information, while \({\lambda }_{{{{\rm{infer}}}}}\) is adjustable during inference to adapt to specific dataset characteristics and annotation priorities (Supplementary Note 3, Supplementary Table S2). This distinction ensures that the model is trained consistently while allowing flexibility in inference to optimize performance based on the dataset’s unique requirements.
MLP decoder
The decoder component of Spot2vector consists of a three-layer MLP framework designed to output parameters for the ZINB distribution. From the low-dimensional representations obtained through the encoder, we used three MLP decoders (\({D}_{\mu }\), \({D}_{\theta }\) and \({D}_{\pi }\)) to generate matrices \({{{\rm{{M}}}}}={({\mu }_{{ij}})}_{S* G}\), \({{\Theta }}={({\theta }_{{ij}})}_{S* G}\) and \({{\Pi }}={({\pi }_{{ij}})}_{S* G}\), representing the expectation, dispersion and zero-inflated probability of the ZINB distribution, respectively51.
The probability distribution function of the negative binomial (NB) distribution is expressed in terms of its mean \({{{\rm{{M}}}}}\) and dispersion \({{\Theta }}\):
The ZINB distribution incorporates zero-inflation probability parameters \({{\Pi }}\) to account for dropout event in gene expression data:
where \({\delta }_{0}\left(x\right)\) is the Dirac delta function, which is equal to 1 if \(x=0\), and 0 otherwise.
The parameters \({{{\rm{{M}}}}}\), \({{\Theta }}\), and \({{\Pi }}\) are inferred through the MLP decoders as follows:
Optimization
The optimization objective of Spot2vector involves minimizing the negative log-likelihood of the ZINB distribution given the ST data \(X\):
where the L1 regularization term \({{||}{{\Theta }}{||}}_{1}\) is added to encourage sparsity in the dispersion parameters \({{\Theta }}\).
Expression recovery
The matrix \({{{\rm{{M}}}}}\) represents the expected counts of expression data. This adjustment removes noise and recovers spatial expression patterns of genes. The denoised expression data helps distinguish true zeros, which occur when certain genes are biologically inactive in specific cell types (e.g., a gene not expressed in a non-relevant cell type), from false zeros that arise due to random variation or technical noise (e.g., low-expressed genes not detected by sequencing technology). Compared to direct denoising by fitting the original matrix, this method can provide stronger capabilities for expression data recovery, and it can also explain the origins of the noise through a mathematical model.
Calculation of Ripley’s K and L functions
Ripley’s \(K(r)\) and \(L(r)\) are used to analyze spatial point distributions for clustering and uniformity. \(K(r)\) reflects the point distribution pattern by calculating the number of point pairs within a radius \(r\). \(L(r)\) is a smooth transformation of \(K(r)\), typically computed as \(L\left(r\right)=\sqrt{K\left(r\right)/\pi }-r\), removing the theoretical effect of uniform distribution. However, in ST data such as 10X Visium, the spatial spots are arranged in a regular hexagonal grid, with a fixed minimum spacing \({d}_{\min }\). This regularity violates the assumption of random spatial distributions underlying the classical \(K(r)\) and \(L(r)\) calculations, necessitating an adjustment to account for the hexagonal arrangement.
To accommodate this structural constraint and ensure accurate spatial analysis, a normalization method is introduced. Given a minimum point distance of \({d}_{\min }\), the theoretical number of point pairs within a radius \(r\) is computed as:
Then, the normalization factor is calculated as:
Finally, \(L(r)\) is adjusted to:
This normalization method effectively removes the influence of the regular hexagonal spacing, providing a more accurate description of the actual spatial structure in ST data.
Adaptive thresholding of gene expression
To identify the expression threshold for separating background and signal, we utilize the density distribution of gene expression values, estimated via Gaussian Kernel Density Estimation (KDE). First, the peaks in the expression density curve are located using a peak detection algorithm. If two or more peaks are identified, the minimum value (trough) between the first two peaks is determined, and its corresponding expression value is used as the threshold, under the assumption that the first peak represents background and the second and subsequent peaks represent signal. If no distinct bimodal distribution is detected, the global median of the expression values is used as a fallback threshold.
This peak-based threshold selection algorithm adapts to scale differences introduced by various denoising algorithms applied to gene expression data, ensuring robust and consistent separation of background and signals.
Implementation
The Spot2vector framework was implemented using PyTorch. The model was trained using the Adam optimizer with a learning rate of \(1\times {10}^{-4}\). Based on empirical validation, the embedding dimension was defaulted to 32, which not only ensures optimal performance but also maintains computational efficiency. This specific dimension adequately captures essential features without incurring excessive resource consumption. To further understand the impact of embedding dimensionality, we evaluated the robustness of Spot2vector’s performance using the MouseBrain dataset (Supplementary Fig. S23). The results indicate that Spot2vector maintains robust ___domain identification performance when the embedding dimensionality varies within a reasonable range. The parameter \({\lambda }_{{{{\rm{train}}}}}\) was set to 0.5 to ensure balanced learning from both spatial and expression information. This equal weighting allows the model to effectively integrate both data types during training.
Unsupervised clustering
After obtaining the embeddings of spots, we can use unsupervised clustering algorithms to identify the ___domain classifications, and compare them with the true ___domain labels. In this study, we use the “mclust” algorithm to perform spatial clustering27, which was developed based on Gaussian Mixture Model (GMM) to infer the latent structure of data. We also conducted additional experiments using three alternative clustering methods: Louvain, Leiden, and Bayesian Gaussian Mixture (BGMM). Our comprehensive comparison revealed that the ___domain identification results were relatively stable across different clustering methods for most datasets, and mclust consistently demonstrated superior clustering performance. (Supplementary Fig. S24). Since the clustering algorithm is an independent downstream analysis step separate from the Spot2vector model, we have integrated Louvain and Leiden into the downstream analysis module of Spot2vector to provide users with more flexible options. By default, mclust remains the recommended clustering method due to its proven performance and robustness.
Statistics and reproducibility
The scripts related to this study are available at https://github.com/amssljc/Spot2vector/tree/main/tutorials. The parameters of the tools utilized have not been specifically optimized. Users are encouraged to download all raw data (https://github.com/amssljc/Spot2vector/tree/main/data) and can freely modify parameters according to the provided scripts.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All datasets analyzed in this study were publicly available (Supplementary Note 1). The human DLPFC datasets were downloaded from the source study at https://github.com/LieberInstitute/spatialLIBD. The raw 10X Visium Human Breast cancer dataset can be downloaded from https://www.10xgenomics.com/datasets/human-breast-cancer-block-a-section-1-1-standard-1-1-0, and its annotation was downloaded from https://huggingface.co/datasets/han-shu/st_datasets/tree/main. For the adult mouse brain 10X Visium dataset, a pre-processed version with ___domain labels was downloaded from https://squidpy.readthedocs.io/en/stable/api/squidpy.datasets.visium_fluo_adata_crop.html#. The AMBA MERFISH data was downloaded from https://gene.ai.tencent.com/SpatialOmics/dataset?datasetID=184. The MOSTA data was downloaded from https://ftp.cngb.org/pub/SciRAID/stomics/STDS0000058/stomics/. More details about the datasets are provided in Supplementary Table S1. The source data of the main figures are at Supplementary Data 1.
Code availability
The Spot2vector software is available at https://github.com/amssljc/Spot2vector.
References
Xu, C. et al. DeepST: identifying spatial domains in spatial transcriptomics by deep learning. Nucleic Acids Res 50, e131–e131 (2022).
Hu, J. et al. SpaGCN: Integrating gene expression, spatial ___location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Dong, K. & Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 13, 1739 (2022).
Ståhl, P.L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792.e21 (2022).
Rodriques, S.G. et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Chen, K.H., Boettiger, A.N., Moffitt, J.R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Moffitt, J.R., Lundberg, E & Heyn, H. The emerging landscape of spatial profiling technologies. Nat. Rev. Genet 23, 741–759 (2022).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Svensson, V., Vento-Tormo, R. & Teichmann, S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 13, 599–604 (2018).
Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023).
Shao, X. et al. Knowledge-graph-based cell-cell communication inference for spatially resolved transcriptomic data with SpaTalk. Nat. Commun. 13, 4429 (2022).
Vickovic, S. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019).
Li, K. et al. Computational elucidation of spatial gene expression variation from spatially resolved transcriptomics data. Mol. Ther. Nucleic Acids 27, 404–411 (2022).
Marx, V. Method of the Year: spatially resolved transcriptomics. Nat. Methods 18, 9–14 (2021).
Wang, Y. et al. Sprod for de-noising spatially resolved transcriptomics data based on position and image information. Nat. Methods 19, 950–958 (2022).
Yu, Q., Jiang, M. & Wu., L. Spatial transcriptomics technology in cancer research. Front Oncol. 12, 1019111 (2022).
Hua, J., Liu, H., Zhang, B. & Jin, SLAK Lasso and K-Means Based Single-Cell RNA-Seq Data Clustering Analysis. IEEE Access 8, 129679–129688 (2020).
Wolf, FA., Angerer, P. & Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 19.15 (2018).
Traag, V.A., Waltman, L. & van Eck, N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Blondel, V.D., Guillaume, J-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Long, Y. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat. Commun. 14, 1155 (2023).
Pham, D. et al. Robust mapping of spatiotemporal trajectories and cell–cell interactions in healthy and diseased tissues. Nat. Commun. 14, 7739 (2023).
Maynard, KR. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 24, 425–436 (2021).
Wu, SZ. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet 53, 1334–1347 (2021).
Ni, Z. et al. SpotClean adjusts for spot swapping in spatial transcriptomics data. Nat. Commun. 13, 2971 (2022).
Luca, S., Michael, F., Murphy, T.B. & Adrian, E.R .mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R. J. 8, 289–317 (2016).
Allen, W.E., Blosser, T.R., Sullivan, Z.A., Dulac, C. & Zhuang, X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 186, 194–208.e18 (2023).
Xu, H. et al. Unsupervised spatially embedded deep representation of spatial transcriptomics. Genome Med 16, 12 (2024).
Li, Y., Duan, Q & Tan, Y. A pan-cancer analysis of MUC family genes as potential biomarkers for immune checkpoint therapy. J. Clin. Oncol. 39, 2598 (2021).
Rao, C.V., Janakiram, N.B. & Mohammed, A. Molecular Pathways: Mucins and Drug Delivery in Cancer. Clin. Cancer Res 23, 1373–1378 (2017).
Zhai, C. et al. The function of ORAOV1/LTO1, a gene that is overexpressed frequently in cancer: essential roles in the function and biogenesis of the ribosome. Oncogene 33, 484–494 (2014).
Yamada, A. et al. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res Treat. 137, 773–782 (2013).
Honorat, M. et al. ABCC11 expression is regulated by estrogen in MCF7 cells, correlated with estrogen receptor α expression in postmenopausal breast tumors and overexpressed in tamoxifen-resistant breast cancer cells. Endocr. Relat. Cancer 15, 125–138 (2008).
Hlaváč, V. et al. Role of Genetic Variation in ABC Transporters in Breast Cancer Prognosis and Therapy Response. Int J. Mol. Sci. 21, 9556 (2020).
Yamada, Y., Yoshimatsu, K., Yokomizo, H., Okayama, S. & Shiozawa, S. Expression of ATP-binding Cassette Transporter 11 (ABCC11) Protein in Colon Cancer. Anticancer Res 40, 5405 (2020).
Ishikawa, T., Toyoda, Y., Yoshiura, K. & Niikawa, N. Pharmacogenetics of human ABC transporter ABCC11: new insights into apocrine gland growth and metabolite secretion. Front Genet 3, 306 (2013).
Chen, L. et al. Characterization of the bovine immunoglobulin lambda light chain constant IGLC genes. Vet. Immunol. Immunopathol. 124, 284–294 (2008).
Wang, J. et al. Functional analysis of tumor-derived immunoglobulin lambda and its interacting proteins in cervical cancer. BMC Cancer 23, 929 (2023).
Chang, Y-T. et al. A Novel IGLC2 Gene Linked with Prognosis of Triple-Negative Breast Cancer. Front Oncol. 11, 759952 (2022).
Pei, G., Lan, Y., Lu, W., Ji, L & Hua, Z.C. The function of FAK/CCDC80/E cadherin pathway in the regulation of B16F10 cell migration. Oncol. Lett. 16, 4761–4767 (2018).
Yu, M., Peng, J., Lu, Y., Li, S. & Ding, K. Silencing immune-infiltrating biomarker CCDC80 inhibits malignant characterization and tumor formation in gastric cancer. BMC Cancer 24, 724 (2024).
Wang, W-D. et al. A prognostic stemness biomarker CCDC80 reveals acquired drug resistance and immune infiltration in colorectal cancer. Clin. Transl. Med. 10, e225 (2020).
Pohl, S., Scott, R., Arfuso, F., Perumal, V. & Dharmarajan, A. Secreted frizzled-related protein 4 and its implications in cancer and apoptosis. Tumor Biol. 36, 143–152 (2015).
Zhang, W. et al. Secreted frizzled-related proteins: A promising therapeutic target for cancer therapy through Wnt signaling inhibition. Biomed. Pharmacother. 166, 115344 (2023).
Lopez, R., Regier, J., Cole, M.B., Jordan, M.I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
van Dijk, D. et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e27 (2018).
Stuart, T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019).
Kipf, T. N. & Welling, M. Semi-supervised classification with graph convolutional networks. In Proc. International Conference on Learning Representations (ICLR). https://doi.org/10.48550/arXiv.1609.02907 (2017).
Veličković, P. et al. Graph attention networks. In Proc. International Conference on Learning Representations (ICLR). https://doi.org/10.48550/arXiv.1710.10903 (2018).
Yu, Z. et al. ZINB-Based Graph Embedding Autoencoder for Single-Cell RNA-Seq Interpretations. Proc. AAAI Conf. Artif. Intell. 36, 4671–4679 (2022).
Acknowledgements
This work was supported by National Key Research and Development Program of China (No. 2022YFB4500300), Key Research Project of Zhejiang Lab (No. 2024SSYS0005) and the Startup Foundation for Introducing Talent of NUIST, China (No. 2024r088). All authors have read and approved the final version.
Author information
Authors and Affiliations
Contributions
Jiacheng Leng: Conceptualization, Methodology, Software. Jiating Yu: Analysis, Writing, Software. Ling-Yun Wu: Writing—Review & Editing. Hongyang Chen: Writing—Review & Editing, Supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Nam D. Nguyen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan, Laura Rodriguez Perez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Leng, J., Yu, J., Wu, LY. et al. Flexible integration of spatial and expression information for precise spot embedding via ZINB-based graph-enhanced autoencoder. Commun Biol 8, 556 (2025). https://doi.org/10.1038/s42003-025-07965-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07965-5
